一种适合HEK293细胞的双顺反子表达载体及其制备方法、表达系统、应用与流程
一种适合hek293细胞的双顺反子表达载体及其制备方法、表达系统、应用,属于基因工程
技术领域:
。
背景技术:
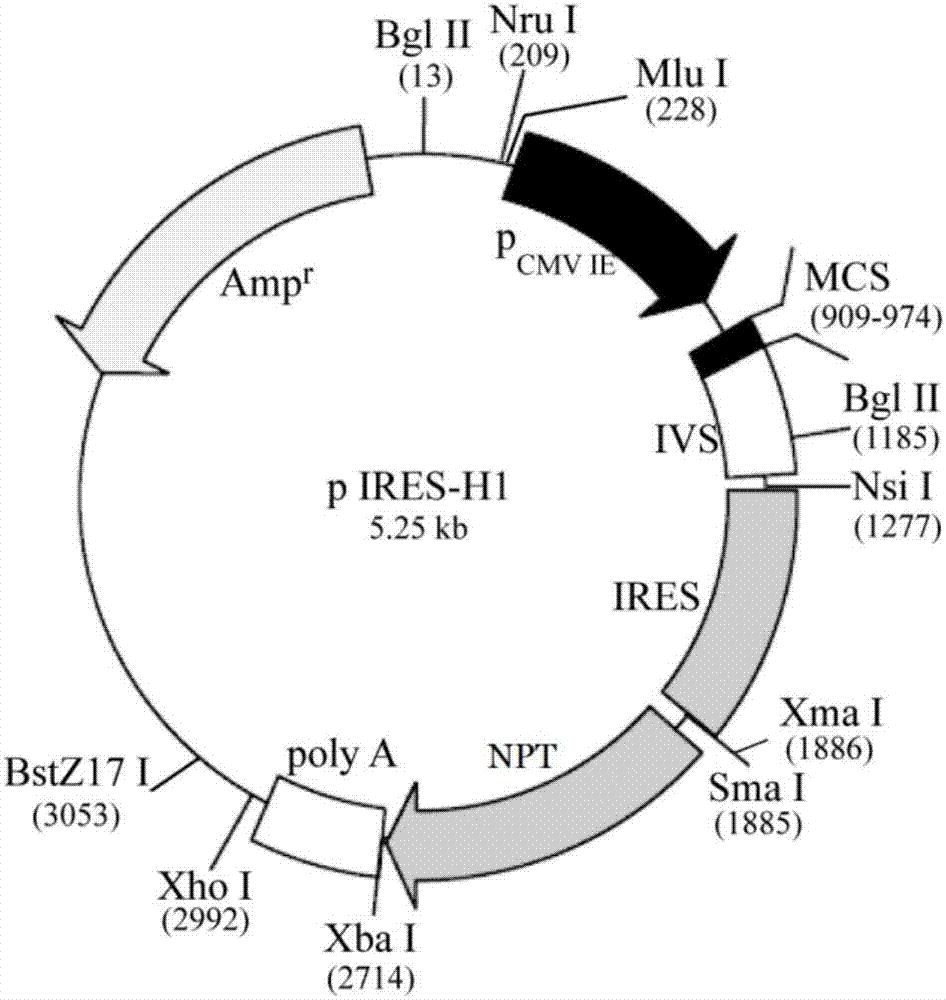
:20世纪70年代基因工程技术的发展,使原来利用动物脏器、组织或人类血液提取蛋白质药物转变为利用基因工程生产。药物蛋白包括单克隆抗体、多肽、蛋白,已经成为医药工业的重要部分,并将成为未来医药发展的重要领域。重组药物蛋白的表达系统主要包括e.coli、酵母以及非人源化哺乳动物细胞系。e.coli适合表达分子量较小、结构相对简单的蛋白质,酵母表达系统适合表达分子量较大、结构较复杂、较少糖基化的蛋白质。结构复杂或糖基化修饰对于蛋白质的活性非常重要,非人源化哺乳动物细胞由于具备类似于人类细胞的翻译后加工修饰(post-translationalmodifications,ptms),因此,非人源化哺乳动物细胞表达系统是目前药物重组药物蛋白生产的重要平台。常用的非人源化哺乳动物细胞主要包括:如中国仓鼠卵巢(chinesehamsterovary,cho)、乳仓鼠胚肾细胞(babyhamsterkidney,bhk)、nso小鼠胸腺瘤细胞、sp2/0骨髓瘤细胞、nso小鼠骨髓瘤细胞。尽管非人源化哺乳动物类细胞表达重组药物蛋白具备安全性和有效性,但是由于ptms方式不同,尤其是糖基化修饰与人源细胞存在差别。如鼠类细胞缺乏人源细胞的糖基化的机制,表达蛋白容易使人体产生免疫性,导致外来的重组药物蛋白被循环系统清除。hek293细胞来源于人胚肾细胞,最早由graham于1977年利用转染腺病毒5型dna获得,近年来被应用于重组药物蛋白和病毒载体的生产。hek293能够在sfm悬浮生长、易转染,具备生产重组药物蛋白的优点。对hek293细胞进行驯化和改造,已经培育出能在sfm生长迅速、转染效率高、表达水平高的细胞系。迄今已有5种由hek293细胞系生产的药物蛋白获美国fda或ema批准生产。hek293细胞表达系统也存在目的蛋白表达水平低、高产稳定细胞筛选周期长、细胞培养成本高等缺陷,这严重制约着重组蛋白药物的生产。而影响重组蛋白在hek293细胞中表达的因素众多,表达载体就是其中一个重要因素。此外,在细胞表达系统中,高水平持续稳定表达的细胞株往往难以得到,这是因为转基因转染细胞后的随机整合,使有些转基因发生沉默或者表达水平降低,因此需要从大量细胞克隆株中分离出稳定高效表达的克隆细胞株,这也给基因工程产业化生产带来了很大困扰。核基质结合区序列(matrixattachmentregion,mar)是限制酶消化后仍附着在核基质上的dna序列,长度为300~3000bp,富含at碱基对(>60%),以及几个短的“共有序列”,如a-box(aataaayaaa)、t-box(ttwtwttwtt)、dna链解旋序列(aatatattt或aatatt)、拓扑异构酶ii结合位点(gtnwayattnatnnr)等。研究表明,mar序列能提高hek293细胞附着体载体(episomalvectors)转基因表达水平,同时降低转化株转基因表达水平差异(modifieds/marepisomalvectorsforstablyexpressingfluorescentprotein-taggedtransgeneswithsmallcell-to-cellfluctuations.analbiochem.2013)。同时研究还发现野猪的mar能提高hek293细胞转基因表达水平(susscrofamatrixattachmentregionenhancesexpressionofthepiggybacsystemtransfectedintohek293tcells.biotechnollett.2016)。已有研究证明,mar提高转基因表达与载体上的其他调控元件(regulatoryelements)有关,如mar与不同启动子组合对提高重组蛋白的表达水平不一等(impactofusingdifferentpromotersandmatrixattachmentregionsonrecombinantproteinexpressionlevelandstabilityinstablytransfectedchocells.molbiotechnol.2014)。公开号为cn102994536a的中国发明专利公开了一种双顺反子供表达基因转移体及其制备方法,公开了从5′端起到3′端止依次包括有牛核基质结合区mar、人工构建的组合启动子cag、抑制蛋白基因fstn、内部核糖体进入位点ires、绿色荧光蛋白acgfp基因、rabbitglobinpolya信号区的表达载体,但是其mar序列来源于牛,不适合人来源的hek293细胞系,并且该表达载体没有包括筛选标记,不能进行稳定转化细胞株的筛选。公开号为cn106520832a的中国发明专利公开了一种双顺反子表达载体,但是由于不同的mar元件组合对转基因的表达也有不同作用(matrixattachmentregioncombinationsincreasetransgeneexpressionintransfectedchinesehamsterovarycells.scirep.2017),载体的骨架结构与其转染的宿主细胞有关(cellcompatibilityofaneposimalvectormediatedbythecharacteristicmotifsofmatrixattachmentregions.currgenether,2016),适合cho细胞表达的载体不一定适合hek293细胞(evaluatingpost-transcriptionalregulatoryelementsforenhancingtransientgeneexpressionlevelsinchok1andhek293cells.proteinexprpurif.2010),因此研发适合hek293细胞的表达载体有重要意义。技术实现要素:本发明的目的在于提供一种适合hek293细胞的双顺反子表达载体。本发明还提供了上述双顺反子表达载体的制备方法。本发明还提供了上述双顺反子表达载体的表达系统。本发明还提供了上述双顺反子表达载体的应用。为了实现上述目的,本发明所采用的技术方案是:一种适合hek293细胞的双顺反子表达载体,包含如下元件:第一核基质结合区序列-启动子-目的基因-内部核糖体进入位点序列-筛选标记基因-polya–第二核基质结合区序列。所述第一核基质结合区序列为人x-29mar序列、人β-珠蛋白mar序列、鸡卵溶菌酶mar序列、人hprtmar序列或mar1-68序列;所述第二核基质结合区序列为人x-29mar序列、人β-珠蛋白mar序列、鸡卵溶菌酶mar序列、人hprtmar序列或mar1-68序列。所述第一核基质结合区序列与第二核基质结合区序列为人x-29mar序列、人β-珠蛋白mar序列、鸡卵溶菌酶mar序列、人hprtmar序列或mar1-68序列中的任意两种。优选的,所述第一核基质结合区序列和第二核基质结合区序列为人来源的核基质结合区序列。优选的,所述第一核基质结合区序列为人x-29mar序列,genbank:ef694970.1,第1~3337位碱基。优选的,所述第二核基质结合区序列为人β-珠蛋白mar序列为genbank:l22754.1,第840~2998位碱基。所述鸡卵溶菌酶mar序列为genbank:x98408,第1~1668位碱基;所述人hprtmar序列,为genbank:x07690,第1~580位碱基。所述mar1-68为genbank:ef694965.1,第1~3614位碱基。所述启动子为sv40启动子、cmv启动子、ef-1α启动子中的一种。优选的,所述启动子为cmv启动子,其核苷酸序列如seqidno.1所示。所述筛选标记基因为新霉素磷酸转移酶基因或博来霉素抗性基因。优选的,所述筛选标记基因为npt抗性弱化基因,其核苷酸序列如seqidno.2所示。所述目的基因为药物重组蛋白基因。优选的,所述目的基因为促红细胞生成素基因。作为本发明的一种实施方式,所述的双顺反子表达载体以pires-neo载体为出发载体构建。所述出发载体为可以根据需求进行改造的初始载体。所述hek293细胞为hek293-h、hek293-f或hek293-t细胞。上述的双顺反子表达载体的构建方法:包括如下步骤:1)将筛选标记基因连接至pires-neo载体上,得pires-h1载体;2)在pires-h1载体的启动子前和polya后分别插入第一核基质结合区序列和第二核基质结合区序列,即得pires-h2载体。步骤1)中所述将筛选标记基因连接至pires-neo载体的具体操作为:采用xmai和xbai酶双酶切seqidno.3所示序列和pires-neo载体,回收酶切片段,连接、转化至大肠杆菌中后鉴定,得到pires-h1载体。步骤2)中所述在pires-h1载体的启动子前和polya后分别插入核基质结合区序列的具体操作为:采用nrui和mlui双酶切合成人x-29mar序列和pires-h1质粒dna,回收酶切片段,连接、转化至大肠杆菌中后鉴定,得pires-h1m载体;采用xhoi和bstz17i双酶切β-珠蛋白mar序列和pires-h1m质粒dna,连接、转化至大肠杆菌中后鉴定,得到pires-h2载体。上述的双顺反子表达载体构建得到的hek293表达系统。上述的hek293表达系统的构建方法:包括:将所述双顺反子表达载体转染进入hek293细胞中,即得。所述转染为磷酸钙法、电转法、脂质体转染法转染。优选的,所述转染为电转法转染。上述的双顺反子表达载体在制备药物蛋白中的应用。本发明提供一种适合hek293细胞的双顺反子表达载体包含核基质结合区序列、内核糖体进入位点序列和筛选标记基因,通过在启动子与ires序列之间插入目的基因,可构成目的基因-ires序列-筛选标记基因依次连接的双顺反子序列。表达载体的结构为:mar-启动子-目的基因-ires序列-筛选标记基因-polya-mar。本发明的载体含有适合hek细胞系表达的mar序列,并且含有弱化的筛选标记,该载体利用优化的mar序列可以显著提高hek293细胞转基因表达,克服转基因沉默,实现转基因在宿主细胞中的高效、长期表达。同时可实现目的基因和筛选标记的同时表达,可减少由于不同表达框存在导致的假阳性细胞克隆,提高阳性细胞克隆筛选率,并克服传统载体目的基因与筛选标记基因表达不均衡的问题。附图说明图1为实施例1中pires-neo载体结构示意图;图2为实施例1中的pires-h1载体结构示意图;图3为实施例1中的pires-h2载体结构示意图;图4为实施例2中epo在hek293细胞单克隆中的表达水平统计图;图5为epo在不同载体稳定筛选的平均表达水平比较图。具体实施方式以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。实施例及试验例中所用大肠杆菌(escherichiacoli)jm109、pires-neo质粒载体、细胞系试剂、工具酶等均为市售商品。内切酶及nebuffer均购自美国newenglandbiolabsltd(neb),pires-neo质粒载体购自clontech生物公司。实施例1适合hek293细胞的双顺反子表达载体的构建,具体步骤如下:1、pires-h1载体的构建本步骤的目的为将pires-neo载体(其结构如图1所示)上的筛选标记基因替换为npt抗性弱化基因。1)人工合成npt抗性弱化基因设计并人工合成npt抗性弱化基因(如seqidno.3所示),具体交由通用生物基因(安徽)有限公司完成。为便于克隆及保证序列完整性,合成npt抗性弱化基因时,5′端引入aaccccgggataattcctgcagccaat序列,其中aac为保护碱基,cccggg为xmai酶切位点,ataattcctgcagccaat为载体pires-neo上的部分序列(genbank:u89673.1,第1892~1909位碱基);3′端引入ggggatcaattctctagagct序列,其中gct为保护碱基,tctaga为xbai酶切位点,ggggatcaattc为载体pires-neo上的部分序列(genbank:u89673.1,第2714~2725位碱基)。2)构建pires-h1载体用xmai/xbai双酶切合成的npt抗性弱化基因,同时用xmai/xbai双酶切pires-neo质粒dna,再用neb公司tm的连接试剂盒25℃连接5min。将连接产物加入到e.colijm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组质粒并进行双酶切(xmai/xbai)验证,取酶切验证正确的质粒进行测序验证,构建正确的质粒命名为pires-h1,其结构示意图如图2所示。2、pires-h2载体的构建本步骤的目的为在pires-h1载体的启动子cmv的上游插入人x-29mar序列及polya的下游插入β-珠蛋白mar序列。1)合成人x-29mar序列根据人x-29mar序列(genbank:ef694970.1,第1~3337位碱基),具体交由通用生物基因(安徽)有限公司完成。为便于克隆,合成人x-29mar序列时,5'端引入gtctcgcga序列,其中gtc为保护碱基,tcgcga为nrui酶切位点。3′端引入agcacgcgt,其中agc作为酶切的保护碱基,acgcgt为mlui酶切位点。2)构建启动子cmv上游含人x-29mar序列的表达载体用nrui/mlui双酶切合成的人x-29mar序列,同时用nrui/mlui双酶切pires-h1质粒dna。琼脂糖凝胶电泳鉴定酶切结果,凝胶回收酶切后的人x-29mar序列片段和pires-h1线形质粒dna。人x-29mar序列的双酶切体系为:人x-29mar序列10μl(1μg/μl),10×nebuffer3.13μl,nrui/mlui(10u/μl)各1.0μl,补足水至30μl;酶切条件为:37℃,酶切3min。pires-h1质粒的双酶切体系为:pires-h1质粒5μl(1μg/μl),10×nebuffer3.12μl,nrui/mlui(10u/μl)各0.5μl,补足水至20μl;酶切条件为:37℃,酶切3min。取酶切后的人x-29mar序列片段和pires-h1线形质粒dna(摩尔比5:1),使用neb公司tm的连接试剂盒,25℃连接5min。将连接产物加入到e.colijm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组质粒并进行双酶切(nrui/mlui)验证,取酶切验证正确的质粒进行测序验证,构建正确的质粒命名为pires-h1m。3)pcr扩增β-珠蛋白mar序列根据β-珠蛋白mar序列(genbank:l22754.1,第840~2998位)设计引物p1、p2、。在引物p1、p2的5′端、3′端分别引入5′端分别引入xhoi、bstz17i酶切位点,引物序列如下(下划线为酶切位点):引物p1:5′-gtcctcgagaatatatctcctgataaaatgtcta-3′(如seqidno.4所示);引物p2:5′-gtcgtatacggatcctcccatttcggcctcctg-3′(如seqidno.5所示)。提取人外周血基因组dna作为模板,分别用引物p1/p2进行pcr扩增,扩增反应体系见下表1。表1pcr扩增反应体系pcr反应体系试剂浓度体积(μl)终浓度ddh2o/17/p1/p210μmol/l各1.00.4μmol/l10×pcrbuffer/2.51×dntp25μmol/l2.0200μmol/l模板dna100ng/μl1.04.0ng/μltaq酶5u/μl0.50.1u/μlpcr扩增程序为:95℃3min,94℃40s,58℃30s,72℃40s,每个退火温度4个循环,最后55℃,30个循环,72℃3min。琼脂糖凝胶电泳回收扩增产物,送生物公司测序验证。结果表明,扩增出的dna片段均与genbank登录的β-珠蛋白mar序列一致。4)构建启动子cmv上游含人x-29mar及polya下游含β-珠蛋白mar序列的表达载体用xhoi/bstz17i双酶切p1/p2扩增出的β-珠蛋白mar序列,同时用xhoi/bstz17i双酶切pires-h1m质粒dna。琼脂糖凝胶电泳鉴定酶切结果,凝胶回收酶切后的β-珠蛋白mar序列片段和pires-h1m线形质粒dna。使用neb公司tm的连接试剂盒,25℃连接5min。将连接产物加入到e.colijm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组质粒并进行双酶切(xhoi/bstz17i)验证,取酶切验证正确的质粒进行测序验证,构建正确的质粒命名为pires-h2,其结构示意图如图3所示。实施例2真核细胞表达系统的构建,具体步骤如下:1、构建含促红细胞生成素(epo)外源基因的双顺反子表达载体1)合成epo序列根据ncbi公布的epo序列(genbank:jn849371.1,第1~582位碱基),人工合成epo序列(5′端和3′端分别引入ecori、bamhi酶切位点),具体交由通用生物基因(安徽)有限公司完成。2)构建含epo序列的双顺反子表达载体用ecori/bamhi双酶切引物人工合成的eop序列,同时用ecori/bamhi双酶切pires-neo及pires-h2质粒dna。琼脂糖凝胶电泳鉴定酶切结果,凝胶回收酶切后的epo序列片段和pires-neo、pires-h2线形质粒dna。epo序列的双酶切体系为:epo序列片段10μl(1μg/μl),10×nebuffer2.13μl,ecori/bamhi酶(10u/μl)各1.0μl,补足水至30μl;酶切条件为:37℃,酶切3min。pires-h2质粒的双酶切体系为:pires-h2质粒5μl(1μg/μl),10×nebuffer2.12μl,ecori/bamhi(10u/μl)各0.5μl,补足水至20μl;酶切条件为:37℃,酶切3min。取酶切后的epo序列片段和pires-h2线形质粒dna(摩尔比5:1),使用neb公司tm的连接试剂盒,25℃连接5min。将连接产物加入到e.colijm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组质粒并进行双酶切(ecori/bamhi)验证,取酶切验证正确的质粒进行测序验证,构建正确的质粒命名为pires-h2-epo。2、构建真核细胞表达系统选择生长状态良好的hek293细胞接种到6孔培养板上,待铺板密度达到约80%进行转染。具体操作步骤如下:10μllipofectamine2000+240μl无血清freestyletm293expressionmedium,在37℃孵箱中静置5min,将无血清培养基与250μl(5μg)表达载体pires-h2-epo混合,37℃孵箱中静置20min;同时用pbs将6孔培养板上的细胞清洗三遍,加入2ml无血清细胞培养基;然后将脂质体与含pires-h2-epo质粒dna的混合液逐滴轻滴入孔中,尽快轻轻摇晃培养平板使其混匀;放入5%co2细胞培养箱中。48h后细胞加入600μg/ml的g418药物,每48h换成新鲜完整无血清培养基,从第五天开始细胞大量死亡。筛选两周后将g418浓度调整到维持浓度300μg/ml继续培养。药物筛选完成后,对细胞进行有限稀释法单克隆化操作。约两个星期单克隆细胞在孔中长至80%,一天后取细胞上清做elisa检测,检测结果见图4。由图4可知,挑选的10个克隆中,1#和4#克隆的产率最佳,分别为256.2mg/l和248.3mg/l。实施例3适合hek293细胞的双顺反子表达载体pires-neo-epo的构建。参照实施例2中方法构建含epo外源基因的pires-neo-epo表达载体,其与pires-h2-epo的区别为:1)筛选标记不同;pires-neo-epo筛选标记为载体上的1906~2709(genbank:u89673.1,第1906~2709位碱基);pires-h2-epo筛选标记序列如seqidno.2所示;2)pires-neo-epo不含有两端的mar序列,pires-h2-epo在cmv启动子前含有人x-29mar序列,在polya的下游含有β-珠蛋白mar序列。构建pires-neo-epo真核细胞表达系统,其构建方法同实施例2。实施例4本实施例中的适合hek293细胞的双顺反子表达载体的构建,包括如下步骤:以pires-neo载体为出发载体,在启动子cmv的上游插入人x-29mar序列,在polya的下游插入人hprtmar序列,插入位点同实施例1。试验例将pires-neo-epo表达载体、pires-h2-epo表达载体以及公开号为cn106520832a的中国发明专利中的pires-c3-epo表达载体使用相同的操作转染至hek293细胞中,获得数量近似的单克隆细胞株,对阳性单克隆群进行elisa检测,并比较克隆株的表达量,各自筛选得到10个稳定细胞株,对10株细胞的epo表达量进行分析。分析结果如图5所示,结果显示,利用本发明表达系统的epo表达水平明显高于传统的表达系统,稳定筛选的epo表达量平均为196.98mg/l,pires-c3-epo表达epo基因表达量达到62.65mg/l,而传统表达系统pires-neo-epo的epo表达量仅为39.45mg/l,本发明的表达系统相对pires-c3-epo提高3.14倍,相对pires-neo-epo提高4.99倍(p<0.01),而公开号为cn106520832a的中国发明专利的pires-c3-epo虽和对照有所提高,但并不显著(仅提高1.59倍)。sequencelisting<110>新乡医学院<120>一种适合hek293细胞的双顺反子表达载体及其制备方法、表达系统、应用<160>5<170>patentinversion3.5<210>1<211>599<212>dna<221>cmv启动子<222>(1)..(599)<400>1agatatacgcgttgacattgattattgactagttattaatagtaatcaattacggggtca60ttagttcatagcccatatatggagttccgcgttacataacttacggtaaatggcccgcct120ggctgaccgcccaacgacccccgcccattgacgtcaataatgacgtatgttcccatagta180acgccaatagggactttccattgacgtcaatgggtggactatttacggtaaactgcccac240ttggcagtacatcaagtgtatcatatgccaagtacgccccctattgacgtcaatgacggt300aaatggcccgcctggcattatgcccagtacatgaccttatgggactttcctacttggcag360tacatctacgtattagtcatcgctattaccatggtgatgcggttttggcagtacatcaat420gggcgtggatagcggtttgactcacggggatttccaagtctccaccccattgacgtcaat480gggagtttgttttggcaccaaaatcaacgggactttccaaaatgtcgtaacaactccgcc540ccattgacgcaaatgggcggtaggcgtgtacggtgggaggtctatataagcagagctct599<210>2<211>795<212>dna<221>npt抗性弱化基因<222>(1)..(795)<400>2atgattgaacaagatggattgcacgcaggttctccggccgcttgggtggagaggctattc60ggctatgactgggcacaacagacaatcggctgctctgatgccgccgtgttccggctgtca120gcgcaggggcgcccggttctttttgtcaagaccgacctgtccggtgccctgaatgaactg180caggacgaggcagcgcggctatcgtggctggccacgacgggcgttccttgcgcagctgtg240ctcgacgttgtcactgaagcgggaagggactggctgctattgggcgaagtgccggggcag300gatctcctgtcatctcaccttgctcctgccgagaaagtatccatcatggctgatgcaatg360cggcggctgcatacgcttgatccggctacctgcccattcgaccaccaagcgaaacatcgc420atcgagcgagcacgtactcggatggaagccggtcttgtcgatcaggatgatctggacgaa480gagcatcaggggctcgcgccagccgaactgttcgccaggctcaaggcgcgcatgcccgac540ggcgatgatctcgtcgtgacccatggcgatgcctgcttgccgaatatcatggtggaaaat600ggccgcttttctggattcatcgactgtggccggctgggtgtggcggaccgctatcaggac660atagcgttggctacccgtgatattgctgaagagcttggcggcgaatgggctgaccgcttc720ctcgtgctttacggtatcgccgctcccgattcgcagcgcatcgccttctatcgccttctt780aacgagttcttctga795<210>3<211>843<212>dna<221>合成npt抗性弱化基因<222>(1)..(843)<400>3aaccccgggataattcctgcagccaatatgattgaacaagatggattgcacgcaggttct60ccggccgcttgggtggagaggctattcggctatgactgggcacaacagacaatcggctgc120tctgatgccgccgtgttccggctgtcagcgcaggggcgcccggttctttttgtcaagacc180gacctgtccggtgccctgaatgaactgcaggacgaggcagcgcggctatcgtggctggcc240acgacgggcgttccttgcgcagctgtgctcgacgttgtcactgaagcgggaagggactgg300ctgctattgggcgaagtgccggggcaggatctcctgtcatctcaccttgctcctgccgag360aaagtatccatcatggctgatgcaatgcggcggctgcatacgcttgatccggctacctgc420ccattcgaccaccaagcgaaacatcgcatcgagcgagcacgtactcggatggaagccggt480cttgtcgatcaggatgatctggacgaagagcatcaggggctcgcgccagccgaactgttc540gccaggctcaaggcgcgcatgcccgacggcgatgatctcgtcgtgacccatggcgatgcc600tgcttgccgaatatcatggtggaaaatggccgcttttctggattcatcgactgtggccgg660ctgggtgtggcggaccgctatcaggacatagcgttggctacccgtgatattgctgaagag720cttggcggcgaatgggctgaccgcttcctcgtgctttacggtatcgccgctcccgattcg780cagcgcatcgccttctatcgccttcttaacgagttcttctgaggggatcaattctctaga840gct843<210>4<211>34<212>dna<213>人工序列<221>引物p1<222>(1)..(34)<400>4gtcctcgagaatatatctcctgataaaatgtcta34<210>5<211>33<212>dna<213>人工序列<221>引物p2<222>(1)..(33)<400>5gtcgtatacggatcctcccatttcggcctcctg33当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1