一种二糖核苷化合物及其制备方法与流程
本发明涉及有机合成技术领域,尤其涉及一种二糖核苷化合物及其制备方法。
背景技术:
二糖核苷是一类天然产物,从自然界已经分离出一百多种二糖核苷(e.v.efimtseva,i.v.kulikova,s.n.mikhailov,cur.org.chem.2007,11,337-354),它们具有抗细菌,杀虫,抗癌,抗病毒等活性(s.n.mikhailov,e.v.efimtseva.collectionsymposiumseries2002,5,181-194),它们是trna和poly(adp-riboes)的片段(e.v.efimtseva,s.n.mikhailov,biochemistry(moscow),2002,67,1136-1144.),可以在核糖的代谢取代过程中生成(k.uchida,y.suzuki,biosci.biotechnol.biochem.2003,67,643-645.)。最近的研究表明,二糖核苷是潜在的parps的抑制剂,有可能成为治疗中风,糖尿病,关节炎等各类炎症,也可能是抗癌和抗病毒的药物(a.s.efremova,a.l.zakharenko,s.i.shram,i.v.kulikova,m.s.drenichev,m.v.sukhanova,s.n.khodyreva,n.f.myasoedov,o.i.lavrik,s.n.mikhailov,nucleosidesnucleotides&nucleicacids,2013,32,510-528)。对二糖核苷合成方法的研究可以为发现parp的抑制剂提供更多的机会。
另外,许多的抗病毒药物,例如hiv药物和hcv药物,都是核苷类化合物,若在它们的糖元部分上加上一个糖,与最初的药物相比,二糖核苷可以提高它们的水溶性及生物利用度,最终达到提高药效的目的(d.benito,m.i.matheu,a.morere,y.diaz,s.castillon,carbohydr.res.,2009,344,2559-2567)。因此,二糖核苷合成方法的研究,在药物研发这个领域将有广范的应用价值。
尽管许多新的合成二糖核苷的方法不断地被发现(y.l.zhang,s.knapp.j.org.chem.,2016,81,2228–2242;a.shin,f.taketo;i.taiki,k.masayuki,s.shigeto,k.shin-ya,chemistry–anasianjournal,2015,10,740-751;e.v.efimtseva,i.v.kulikova,s.nmikhailov,molecularbiology,2009,43,301-312),但与2-去氧呋喃型核糖相关的二糖核苷的合成却未见报道。
因此,现有技术还有待发展。
技术实现要素:
鉴于上述现有技术的不足之处,本发明的目的在于提供一种二糖核苷化合物及其制备方法,旨在为2-去氧呋喃型核糖相关的二糖核苷此类化合物的合成提供可行的方法。
为了达到上述目的,本发明采取了以下技术方案:
一种如通式1所示的二糖核苷化合物,
其中,第一核糖为2-脱氧呋喃型核糖,第二核糖为呋喃型核糖,第一核糖与第二核糖之间由β构型相连;r1、r2、r3、r4、r5为不同的取代基团、r为杂环。
所述的二糖核苷化合物,其中,所述r1、r2、r3、r4、r5分别为不同烷基,芳基,酰基,环烷基及磷酯基。
所述的二糖核苷化合物,其中,r2和r3为h,oh,f或ch3。
所述的二糖核苷化合物,其中,r1、r4、r5为烷基,芳基,酯基,硅烷基,磷脂基或酰基。
所述的二糖核苷化合物,其中,r为保护或者非保护的杂环类基团,与核糖的c1相连的为氮原子,硫原子,氧原子或碳原子。
一种结构通式1所示的二糖核苷化合物的制备方法,其中:
a、将胸腺嘧啶核苷溶解在吡啶中,加入咪唑和叔丁基二苯基氯硅烷,在保护气氛下反应完毕后,以预定的分离步骤获得结构式1所示的化合物;
b、将结构式1所示的化合物溶解在干燥的乙腈中,在保护气氛下,加入三乙胺和三甲基硅氯反应预定时间后,加入三氯氧磷和三氮唑并搅拌;反应完成后,加入水中止反应后,以预定的分离步骤获得结构式2所示的化合物;
c、将结构式2所示的化合物溶解于干燥的dmf中,加入苯甲酸酐,在保护气氛下反应完毕后,以预定的分离步骤获得结构式3所示的化合物;
d、将甲基呋喃型核糖核苷溶解于无水吡啶中,加入乙酸酐进行反应,反应完毕后,以预定的分离步骤获得结构式4所示的化合物;
e、将结构式4所示的化合物溶解于无水1,2-二氧乙烷中,加入sncl4以及结构式3所示的化合物,在保护气氛下反应完毕后,以预定的分离步骤获得结构式5所示的混合物;所述混合物为一对端基差向异构体;
f、将结构式5所示的混合物溶解于四氢呋喃和甲醇混合液里,并加入碳酸钾搅拌反应完毕后,以预定的分离步骤获得结构式6所示的混合物;
g、将结构式6所示的混合物溶解于四氢呋喃并加入四丁基氟化胺,搅拌反应完毕后,以预定的分离步骤获得结构式7所示的混合物;
h、将结构式7所示的混合物溶解在干燥的吡啶中,加入dmap和二对甲氧基苯基苯基甲基氯,搅拌反应完毕后,以预定的分离步骤获得dmt保护的二糖核苷端基差向异构体组成的混合物;
i、通过高压液相分离所述二糖核苷端基差向异构体组成的混合物,形成包含对应的,如结构式8所示的端基差向异构体的复合溶液1和复合溶液2;
j、分别对复合溶液1和复合溶液2进行处理,获得成品。
所述的制备方法,其中,所述预定的分离步骤具体包括:
在反应完毕后,去除反应体系的溶剂;
将反应体系内的残余物通过柱层析分离。
所述的制备方法,其中,所述步骤j具体包括:
分别将复合溶液1和复合溶液2在无水dmf中加入bz2o,室温下搅拌过夜,分别获得如结构式9所示的端基差向异构化合物α和端基差向异构化合物β;
分别将端基差向异构化合物α和端基差向异构化合物β在四氢呋喃和甲醇的混合物中加入饱和碳酸钾溶液,室温下搅拌反应并通过薄层色谱监测;
去除溶剂后溶解在乙酸乙酯中,使用水冲洗有机层后,蒸发并对残留物通过hplc纯化,分别获得包含如结构式10所示的端基差向异构化合物α和端基差向异构化合物β的纯化产物1和纯化产物2;
所述的制备方法,其中,所述步骤j具体包括:
将所述纯化产物1在dmf中共蒸发无水甲苯,其次在氮气和室温下加入tetraisopropylphosphorodiamidite和四唑,反应后加入n-甲基咪唑,在室温下搅拌过夜,经过高效液相色谱纯化获得如结构式11所示的xlvi-5-me-dcamidite;
所述的制备方法,其中,所述通过薄层色谱监测具体包括:添加ph为6的柠檬酸水溶液中和。
有益效果:本发明提供的制备方法可以合成单一2-去氧呋喃型核糖二糖核苷的方法,基于该单一2-去氧呋喃型核糖二糖核苷,可以进一步合成这一类的其它化合物,有利于相关药物的研究或者应用。
附图说明
图1为本发明实施例提供的步骤1的化学反应方程示意图;
图2为本发明实施例提供的步骤2的化学反应方程示意图;
图3为本发明实施例提供的步骤3的化学反应方程示意图;
图4为本发明实施例提供的步骤4的化学反应方程示意图;
图5为本发明实施例提供的步骤5的化学反应方程示意图;
图6为本发明实施例提供的步骤6的化学反应方程示意图;
图7为本发明实施例提供的步骤7的化学反应方程示意图;
图8为本发明实施例提供的步骤8的化学反应方程示意图;
图9为本发明实施例提供的步骤9-1的化学反应方程示意图;
图10为本发明实施例提供的步骤10-1的化学反应方程示意图;
图11为本发明实施例提供的步骤9-2的化学反应方程示意图;
图12为本发明实施例提供的步骤10-2的化学反应方程示意图;
图13为本发明实施例提供的步骤11的化学反应方程示意图;
图14为本发明实施例提供的二糖核苷化合物的结构通式。
具体实施方式
本发明提供一种二糖核苷化合物及其制备方法。为使本发明的目的、技术方案及效果更加清楚、明确,以下参照附图并举实施例对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本发明实施例提供了一种如图14所示的二糖核苷化合物,其中,在图14所示的化合物通式中,第一核糖为2-脱氧呋喃型核糖,第二核糖为呋喃型核糖,第一核糖与第二核糖之间由β构型相连;r1、r2、r3、r4、r5为不同的取代基团、r为杂环,与第一核糖的c1碳连接。
可选地,通式1中的r1、r2、r3、r4、r5分别可以为不同烷基,芳基,酰基,环烷基及磷酯基。另外,通式1中的r可以是保护或者非保护的杂环类基团。r的杂环类集团中,与核糖的c1位置相连的可以为氮原子,硫原子,氧原子或碳原子。
在一些实施例中,r2和r3为h,oh,f或ch3中的一种。而r1、r4、r5为烷基,芳基,酯基,硅烷基,磷脂基或酰基中的一种。其中,r1、r2、r3、r4、r5均可以独立的选择。
通过以下实施例详细陈述上述通式1所示的化合物的合成制备过程。在实施例是图14的通式中其中一个化合物的合成过程。本领域技术人员可以根据实施例提供的详细合成步骤,通过改变使用的原料或者增加基团的取代、置换等步骤可以确定图14的通式所示的其它化合物的合成过程。
在本发明实施例提供的制备方法中,包括如下步骤:
1)将thymidine溶在吡啶中,加入咪唑和叔丁基二苯基氯硅烷。上述反应物在氮气的保护下,搅拌12小时后,加水终止反应。最后,将溶剂蒸干,得到残余物经柱层析分离,得到化合物d1(如图1所示)。
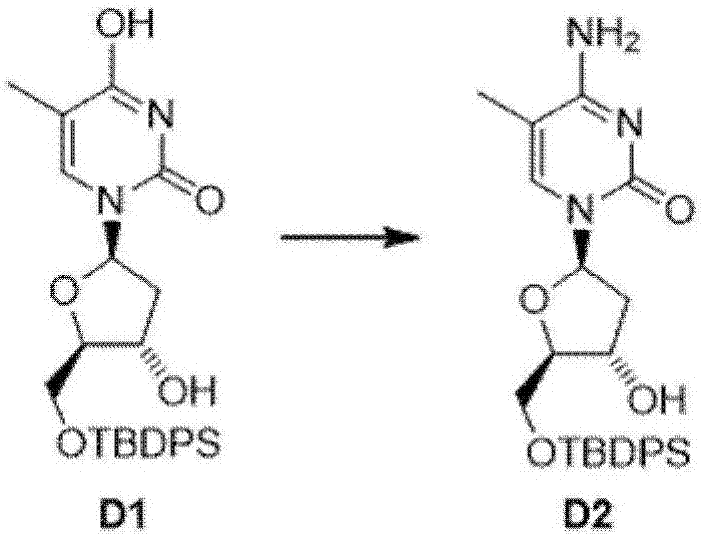
2)将所述化合物d1溶于干燥的乙腈中,在-15℃,在氮气保护的反应条件下,加入三乙胺和三甲基硅氯,反应一小时。然后,在-15℃加入三氯氧磷和三氮唑,并搅拌2小时后,加入水中止反应(如图2所示)。
使用乙酸乙酯萃取,并用无水硫酸钠干燥有机层,减压蒸去溶剂后,将得到的残余物溶于1,4二氧六环和氨水的混合物,在氮水的保护下,室温搅拌12小时,减压蒸去溶剂后,用硅胶柱层析分离残余物(ch2cl2:ch3oh=10:1),得到化合物d2。
3)将化合物d2溶于干燥的dmf中,加入苯甲酸酐。在氮气的保护下,室温搅拌2小时后,将溶剂蒸干,并使用乙酸乙酯溶解残余物。水洗后,用无水硫酸钠干燥,过滤,并将溶剂蒸干后通过硅胶柱层析,得到化合物d3(如图3所示)。
4)将甲基呋喃型核糖糖苷溶于干燥的无水吡啶中,加入乙酸酐,搅拌2小时后,将溶剂蒸干并用硅胶拄分离残余物,得到化合物d4(如图4所示)。
5)化合物d4溶于无水1,2-二氯乙烷,加入sncl4,搅拌15分钟后,加入化合物d3,在0℃和氮气的保护下,搅拌2小时。然后,加入碳酸氢钠水溶液,用二氯甲烷萃取,有机层用无水硫酸钠干燥,过滤,将溶剂蒸干,残余物用硅胶柱层析分离,得到由一对端基差向异物体组成的混合物d5。这对端基差向异构体在各种分离条件下均很难以实现分离,在后续步骤中,直接对这对异构体进行处理(如图5所示)。
6)将混合物d5溶于四氢呋喃和甲醇混合液里,并加入碳酸钾,混合物在室温下搅拌一夜,将溶剂蒸干,残余物用硅胶柱层析分离,得到混合物d6。该混合物d6与混合物d5相类似的,也是由一对端基差向异构体组成(如图6所示)。
7)混合物d6溶于四氢呋喃中,并加入四丁基氟化胺,在室温下搅拌一夜。蒸干液体后,将残留物用硅胶柱层析分离。得到脱保护的二糖核苷的混合物d7(如图7所示)。
8)将混合物d7溶于干燥的吡啶中,加入dmap和二对甲氧基苯基苯基甲基氯,在室温下搅拌4天,蒸去溶剂,残留物用硅胶柱层析分离,得到dmt保护的二糖核苷端基差向异构体的混合物(如图7所示)。此时,这对端基差向异构体可以通过高效液相制备分离,从而获得两个纯的端基差向异构体d8。
9-1)将其中一个端基差向异构体d8的复合溶液(1.4g,1.45mmol)在无水dmf中(9毫升)加入bz2o(0.64克,2.9mmol)。该混合物在氮气下室温搅拌过夜(如图9所示)。
10-1)进一步将步骤9-1)获得的产物在四氢呋喃和甲醇的混合物(1:1,100毫升)加入80毫升饱和碳酸钾溶液。混合物在室温下搅拌反应,并用薄层色谱法进行监测。然后,除去溶剂,残留的溶解在乙酸乙酯(10毫升)中。有机层水冲洗(5毫升×2),蒸发,残渣进行hplc纯化制备0.54克xlvi-5-me-dcpns(如图10所示)。
在本步骤进行所述薄层色谱检测时,材料几乎消失,在一些实施例中,通过添加柠檬酸水溶液(ph=6)中和来解决材料消失,无法进行色谱检测的问题。
9-2)将另一个端基差向异构体d8的复合溶液(1.3g,1.35mmol)在无水dmf(9毫升)加入bz2o(0.60克,2.7mmol)。该混合物在氮气下室温搅拌过夜(如图11所示)。
10-2)进一步将步骤9-2)获得的产物在四氢呋喃和甲醇的混合物(1:1,50毫升)加入80毫升饱和碳酸钾溶液。该混合物在室温下搅拌,并通过薄层色谱法监测。用乙酸乙酯(10毫升)溶解残留物,有机层用水冲洗(5毫升*2),浓缩,最后通过制备型高效液相色谱法对其进行纯化(如图12所示)。
与步骤10-1相类似的,存在进行所述薄层色谱检测时,材料几乎消失的问题。具体可以通过添加柠檬酸水溶液(ph=5)中和来解决材料消失,无法进行色谱检测的问题。
11)如图13所示,可以将在步骤10-1)中获得的xlvi-5-me-dcpns溶液(200毫克,0.188毫摩尔)在dmf1ml共蒸发无水甲苯(3毫升×2),然后在氮气和室温下加入tetraisopropylphosphorodiamidite(452毫克,1.5毫摩尔)和四唑(1.25毫升,0.45m的mecn,0.56毫摩尔)。5分钟后,加入30μl的n-甲基咪唑,和混合物在室温下搅拌过夜。获得的粗产物经高效液相色谱纯化得到性状为白色泡沫的xlvi-5-me-dcamidite。(在本实施例中,最终获得成品150mg,收率为63%)。
可以理解的是,对本领域普通技术人员来说,可以根据本发明的技术方案及本发明构思加以等同替换或改变,而所有这些改变或替换都应属于本发明所附的权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!