从钩吻中分离提纯钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子的方法与流程
本发明涉及一种从钩吻中提取生物碱的方法,特别涉及从钩吻(gelsemiumelegansbenth.)中同时分离提纯钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子的方法,属于天然药物提取领域。
背景技术:
钩吻为马钱科植物胡蔓藤的全草,又名断肠草、野葛、毒根、大茶药等。味苦、辛微、性热,有大毒。主要分布在浙江、福建、广东、广西、湖南、贵州和云南等地。在传统中药常用来治疗类风湿性关节炎疼痛、神经性疼痛、皮肤溃疡等。在兽用方面,少剂量钩吻可对一些动物有促生长的作用。钩吻中主要富含吲哚类生物碱。目前已发现120余种,其中一些生物碱已被证实具有良好的抗肿瘤、抗炎镇痛、免疫作用等活性。在临床上,也常被用来抑制肝肿瘤细胞生长、制备治疗类风湿性关节炎药物、焦虑症药物、慢性疼痛药物等。因此钩吻中的生物碱是其药理活性的一类重要成分。钩吻素子是钩吻中含量最高但毒性较低的一类生物碱,其药理活性上可治疗神经性疼痛、乙酯神经胶质瘤细胞u251,并具有诱导凋亡作用,同时还具有抗应激作用等。钩吻素甲在钩吻中分离到的生物碱含量仅次于素子,能抑制肿瘤细胞生长,并在慢性疼痛中表现为强有效的镇痛作用。目前公开的钩吻素子、素甲的制备方法除使用高速逆流色谱从钩吻总碱中进行制备;还有在钩吻醇提物中用乙酸乙酯进行萃取后,再用硅胶柱层析对其进行分离纯化。高速逆流色谱方法所需设备昂贵,得率低,耗材多。乙酸乙酯方法可能只能萃取得到钩吻吲哚类生物碱中的某一类或几类,其他生物碱可能并未被萃取得到,故不能分离到更多其他生物碱。钩吻碱戊和钩吻绿碱是钩吻中分离得到的两个吲哚类生物碱,其药理作用有抑制肿瘤细胞生长、镇痛、镇静等作用。目前,未出现钩吻碱戊和钩吻绿碱的制备工艺的相关报道。因此,其分离手段的不足是限制其医药用途的一个主要原因。
钩吻成分复杂,活性成分分离以钩吻素甲和钩吻素子为主。目前仅有少数企业对钩吻的某一单一成分进行分离,其他成分做废弃物处理,造成资源利用的不合理,并且仅对钩吻原材料进行某一单体或总碱的分离有报道,但对于钩吻作为原材料进行对多种生物碱的同时提取、分离和纯化,也未见报道。
技术实现要素:
针对现有技术对钩吻中的药物活性成分的分离存在不能同时分离多种药物成分,造成有效成分的浪费,且已分离的单体工艺各有不同,得到的有效成分收率低等缺陷。本发明的目的在于提供一种从钩吻中同时提取分离高纯度钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子的方法,该方法操作简单,成本低,实现了自然资源的综合利用。
为了实现上述技术目的,本发明提供了一种从钩吻中分离提纯钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子的方法,该方法包括以下步骤:
1)将钩吻粉碎后,依次进行酸水提取、中和及干燥,得到酸提取物;所述酸提取物依次进行有机溶剂提取、沉降及离心分离,得到浸膏i,所述浸膏i经过热水溶解、冷冻干燥,得到粉末;
2)所述粉末采用硅胶柱分离,硅胶柱分离过程中以二氯甲烷-甲醇溶液为洗脱剂进行梯度洗脱,依次分别收集含相同成分的馏分,并减压蒸馏,得到含钩吻绿碱的浸膏ii、含钩吻碱戊的浸膏iii、含钩吻素子的浸膏iv、含钩吻素甲的浸膏v和含呋喃钩吻素子的浸膏vi;
3)所述浸膏ii采用硅胶柱纯化和凝胶柱脱色,得到钩吻绿碱;硅胶柱纯化过程中以二氯甲烷-甲醇-氨水溶液为洗脱剂,凝胶柱脱色过程以甲醇为洗脱剂;
4)所述浸膏iii采用硅胶柱纯化,得到钩吻碱戊;硅胶柱纯化过程中以二氯甲烷-甲醇-氨水溶液为洗脱剂;
5)所述浸膏iv采用硅胶柱纯化,得到钩吻素子;硅胶柱纯化过程中以二氯甲烷-甲醇-氨水溶液为洗脱剂;
6)所述浸膏v采用硅胶柱纯化,得到钩吻素甲;硅胶柱纯化过程中以二氯甲烷-甲醇-氨水溶液为洗脱剂;
7)所述浸膏vi采用硅胶柱纯化,得到呋喃钩吻素子;硅胶柱纯化过程中以二氯甲烷-甲醇-氨水溶液为洗脱剂。
优选的方案,所述酸水提过取过程为:固液比为1:6~10kg/l,提取液为无机酸溶液,提取时间为20~28h,提取次数为2~3次。所述无机酸浓度在0.2~0.8wt%范围内。所述无机酸为盐酸、硫酸、磷酸中至少一种;最优选为硫酸溶液。
优选的方案,所述有机溶剂提取过程为:固液比为1:1~2kg/l,提取液为甲酸、乙醇、甲醇、二氯甲烷、乙二醇、正己烷、石油醚中至少一种;提取温度为回流温度,提取时间为1~3h,提取次数为2~3次。提取液优选为浓度为85~95wt%的乙醇;最优选为浓度为90wt%。
优选的方案,所述浸膏i采用45~55℃热水溶解后,在-20℃以下温度下冷冻干燥24~48h得到干燥粉末。
较优选的方案,步骤2)的梯度洗脱过程中在二氯甲烷-甲醇体积比为9:1时,依次收集到含钩吻绿碱、钩吻碱戊、钩吻素子和钩吻素甲馏分,在二氯甲烷-甲醇体积比为7:3时,收集含呋喃钩吻素子的馏分。在其它比例的二氯甲烷-甲醇体系中难以分离得到上述含钩吻绿碱、钩吻碱戊、钩吻素子和钩吻素甲或呋喃钩吻素子的馏分。
较优选的方案,步骤3)中,所述浸膏ii先采用40*600mm硅胶柱,以体积比为95:5:1/20~90:10:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻绿碱的馏分,减压蒸馏,得浸膏ii-1;所述浸膏ii-1采用25*500mm硅胶柱,以体积比为20:0.8:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻绿碱的馏分,减压蒸馏,得浸膏ii-2;所述浸膏ii-2采用20*1800mm凝胶柱,以纯甲醇洗脱剂,收集含钩吻绿碱的馏分,减压蒸馏,得浸膏ii-3;所述浸膏ii-3采用10*400mm硅胶柱,以体积比为19:0.7:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻绿碱的馏分,减压蒸馏,得纯度在98%以上的钩吻绿碱。
较优选的方案,步骤4)中,所述浸膏iii先采用40*600mm硅胶柱,以体积比为95:5:1/20~90:10:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻碱戊的馏分,减压蒸馏,得浸膏iii-1;所述浸膏iii-1采用25*500mm硅胶柱,以体积比为20:0.8:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻碱戊的馏分,减压蒸馏,得纯度在96%以上的钩吻碱戊。
较优选的方案,步骤5)中,所述浸膏iv先采用40*600mm硅胶柱,以体积比为95:5:1/20~90:10:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻素子的馏分,减压蒸馏,得浸膏iv-1;所述浸膏iv-1采用25*500mm硅胶柱,以体积比为20:0.8:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻素子的馏分,减压蒸馏,得纯度在98%以上的钩吻素子。
较优选的方案,步骤6)中,所述浸膏v采用40*600mm硅胶柱,以体积比为95:5:1/20~90:10:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含钩吻素甲的馏分,减压蒸馏,得纯度在92%以上的钩吻素甲。
较优选的方案,步骤7)中,所述浸膏vi先采用40*600mm硅胶柱,以体积比为95:5:1/20的二氯甲烷-甲醇-氨水溶液为洗脱剂进行洗脱,收集含呋喃钩吻素子的馏分,减压蒸馏,得纯度93%呋喃钩吻素子。
本发明的技术方案中中和过程采用碱中和酸水提取溶液至中性。碱为氢氧化钠和/或氢氧化钾。最优为氢氧化钠;氢氧化钠溶液浓度为8mol/l。
本发明的技术方案中二氯甲烷-甲醇系统进行梯度洗脱的过程是二氯甲烷-甲醇按体积比从100:0到0:100进行梯度洗脱。本发明的二氯甲烷和甲醇作为展开剂是经过大量实验优化得到的最佳展开剂组合,如果采用其他组分替换二氯甲烷或甲醇均达不到分离效果。如常规的石油醚和丙酮也无法展开。本发明的洗脱剂采用二氯甲烷-甲醇-氨水溶液作为洗脱剂,适量氨水能有效解决分离过程中化合物拖尾问题。
本发明的凝胶柱采用纯甲醇展开剂,能确保凝胶柱的反复利用。
相对现有技术,本发明的技术方案带来的有益效果:现有技术中的钩吻提取过程中一般仅能提取分离少数种有效组分,造成资源浪费,如钩吻酸水提取物即为工业提取钩吻有效组分后的废弃物。本发明的技术方案,以钩吻为原料,获得酸水提取物,并通过简单工艺同时分离出高纯度的钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子,其中,钩吻绿碱纯度达到98%,而其他组分的纯度均达到92%以上,使天然钩吻资源得到综合利用,且提取分离的方法操作简单,成本低。
附图说明
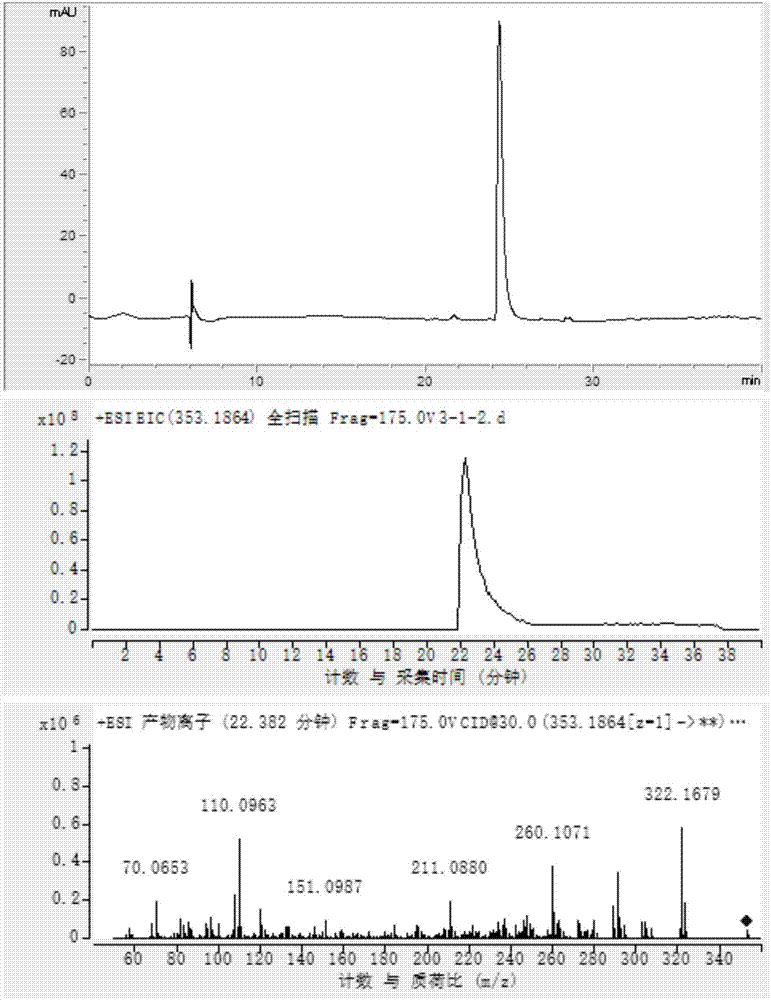
【图1】分别为制得的钩吻绿碱的hplc图谱、eic图、二级图谱;
【图2】为制得的钩吻碱戊的hplc图谱、eic图、二级图谱;
【图3】为制得的钩吻素子的hplc图谱、eic图、二级图谱;
【图4】为制得的钩吻素甲的hplc图谱、eic图、二级图谱;
【图5】为制得的呋喃钩吻素子的hplc图谱、eic图、二级图谱;
【图6】为采用石油醚-丙酮做洗脱剂,3:1点板效果图;
【图7】为本发明的工艺流程图。
具体实施方式
以下实施例旨在进一步说明本发明内容,而不是限制本发明权利要求的保护范围。
实施例1
提取:将100kg粉碎的钩吻加800l的0.5%的硫酸溶液,于容量为1000l的耐酸提取罐中搅拌提取24h,药液用200目滤网过滤,药渣再同法提取1次,合并两次提取液,用氢氧化钠(8mol/l)调ph至中性,然后放于浓缩罐中浓缩至相对密度1.2左右,减压干燥后即得。称取钩吻酸水提取物1kg,投入5l圆底蒸馏瓶,加入90%乙醇回流提取2次,每次均加1.5l,提取2h,提取液液经纱布简单过滤后,收集两次提取液,合并后再放置沉降2h,再通过离心机离心后得离心液,浓缩离心液得浸膏,加入45-55℃热水溶解后,-20℃冷冻干燥48h,得138g干燥粉末。
分离:将138g干燥粉末,加入280g200-300目硅胶拌样,加入已装1.5kg200-300目硅胶的色谱柱中。加入二氯甲烷-甲醇(100:0-0:100)系统梯度洗脱。将洗脱液薄层硅胶板(gf254)点板后于紫外灯254nm下观察,合并单个相同成分馏分。依次得到含钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子洗脱馏分。
纯化:分别浓缩钩吻绿碱、钩吻碱戊、钩吻素子、钩吻素甲和呋喃钩吻素子洗脱馏分。含有钩吻绿碱馏分(展开剂二氯甲烷:甲醇为9:1),减压回收溶剂,得浸膏a;所得浸膏a选用40*600mm硅胶柱,以二氯甲烷:甲醇:氨水体积比为95:5:1/20-90:10:1/20为洗脱剂进行洗脱;经薄层点板后,合并含有钩吻绿碱的馏分,减压回收溶剂,得浸膏b;所得浸膏b选用25*500mm的硅胶柱,以二氯甲烷:甲醇:氨水体积比20:0.8:1/20为洗脱剂进行洗脱,经薄层点板后,合并含有钩吻绿碱的馏分,减压回收溶剂,得浸膏c;(3凝胶色谱纯化)所得浸膏c再用20*1800mm凝胶柱进行脱色,以纯甲醇为展开剂收集洗脱液,经薄层点板后,合并含有钩吻绿碱的馏分,减压回收溶剂,得浸膏d;所得浸膏d选用10*400mm的硅胶柱,二氯甲烷:甲醇:氨水体积19:0.7:1/20;为洗脱剂进行洗脱,薄层点板后得,即得纯化的钩吻绿碱(纯度98%)。
含有钩吻碱戊的馏分(展开剂二氯甲烷:甲醇为9:1),减压回收溶剂,得浸膏a;所得浸膏a选用40*600mm硅胶柱,以二氯甲烷:甲醇:氨水体积比为95:5:1/20-90:10:1/20为洗脱剂进行洗脱;经薄层点板后,合并含有钩吻碱戊的馏分,减压回收溶剂,得浸膏b;所得浸膏b选用25*500mm的硅胶柱,以二氯甲烷:甲醇:氨水体积比20:0.8:1/20为洗脱剂进行洗脱,经薄层点板后,合并含有钩吻碱戊的馏分,减压回收溶剂,即得纯化的钩吻碱戊(纯度为96%)。
含有钩吻素子的馏分(展开剂二氯甲烷:甲醇为9:1),减压回收溶剂,得浸膏a;所得浸膏a选用40*600mm硅胶柱,以二氯甲烷:甲醇:氨水体积比为95:5:1/20-90:10:1/20为洗脱剂进行洗脱;经薄层点板后,合并含有钩吻素子的馏分,减压回收溶剂,得浸膏b;所得浸膏b选用25*500mm的硅胶柱,以二氯甲烷:甲醇:氨水体积比20:0.8:1/20为洗脱剂进行洗脱,经薄层点板后,合并含有钩吻素子的馏分,减压回收溶剂,得纯化的钩吻素子(纯度98%)。
含有钩吻素甲的馏分(展开剂二氯甲烷:甲醇为9:1),减压回收溶剂,得浸膏a;所得浸膏a选用40*600mm硅胶柱,以二氯甲烷:甲醇:氨水体积比为95:5:1/20-90:10:1/20为洗脱剂进行洗脱;经薄层点板后,合并含有钩吻素甲的馏分,减压回收溶剂,得纯化的钩吻素甲(纯度92%)。
含有呋喃钩吻素子的馏分(展开剂二氯甲烷:甲醇为7:3),减压回收溶剂,得浸膏a;所得浸膏a选用40*600mm硅胶柱,以二氯甲烷:甲醇:氨水体积比为95:5:1/20为洗脱剂进行洗脱;经薄层点板后,合并含有呋喃钩吻素子的馏分,减压回收溶剂,即得纯化的呋喃钩吻素子(纯度为93%)。
对比实施例1
提取:将100kg粉碎的钩吻加800l的0.5%的硫酸溶液,于容量为1000l的耐酸提取罐中搅拌提取24h,药液用200目滤网过滤,药渣再同法提取1次,合并两次提取液,用氢氧化钠(8mol/l)调ph至中性,然后放于浓缩罐中浓缩至相对密度1.2左右,减压干燥后即得。称取钩吻酸水提取物1kg,投入5l圆底蒸馏瓶,加入90%乙醇回流提取2次,每次均加1.5l,提取2h,提取液经纱布简单过滤后,收集两次提取液,合并后再放置沉降2h,再通过离心机离心后得离心液,浓缩离心液得浸膏,加入45-55℃热水溶解后,-20℃冷冻干燥48h,得138g干燥粉末。
分离:将138g干燥粉末,加入280g200-300目硅胶拌样,加入已装1.5kg200-300目硅胶的色谱柱中。加入石油醚-丙酮(4:1-1:1)系统梯度洗脱。将洗脱液薄层硅胶板(gf254)点板后于紫外灯254nm下观察,各个馏分在该洗脱剂的比例不能进行很好的分离,未能得到与实施例中一致的成分。
- 还没有人留言评论。精彩留言会获得点赞!