利奥西呱的制备方法与流程
本发明属于药物合成领域,具体阐述了一种制备抗血栓栓塞性疾病药物利奥西呱(riociguat)的新工艺。
背景技术:
利奥西呱,英文名riociguat,中文名:4,6-二氨基-2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基(甲基)氨基甲酸甲酯。该药品由拜耳公司开发,主要用于治疗慢性血栓栓塞性肺动脉高压,已于2013年获批上市。除了拜耳公司对la的研究外,近些年国内对la的关注度也日趋提高,先后公布了cn104530044,cn105294686,cn1048924591,cn105367567,cn105367568,cn105367569等专利。la结构式如下:
在拜耳公司公布的合成路线中,以2-溴苄基氟苯为初始原料经10步反应最终得到目标产品利奥西呱。详细的工艺参数公布在专利wo03095451中,具体如下:
在整个工艺路线中,以2-溴苄基氟苯为初始原料,历经10步反应,最终生成利奥西呱。在由化合物8制备化合物9时,使用了雷尼镍作为催化剂还原制备化合物9,这在一定程度上降低了工艺的安全性,也阻碍了产品的产业化。同时,氢化反应的还原程度也对整体工艺有着重要影响。当还原程度过高时,化合物9继续参与还原反应,从而产生其他杂质,这些杂质会一直带入到终产品中,难以除去。除此之外,化合物8的嘧啶环上存在着三个氨基,虽然以中间的5号位的氨基活性最强,但在后续的酰胺化与甲基化中仍然存在着发生副反应的可能,这也将产生一系列与终产品结构相似的杂质,为后续的纯化带来了很大困难。
随后,拜耳公司又公布了一条新颖的工艺路线,具体内容公布于国际专利wo2012028647和wo2011147810等相关文献。
该工艺的新颖之处在于其嘧啶环由取代法直接引入。在得到中间体17后,继续进行酰胺化与甲基化最终生成利奥西呱。此外,偶联反应带入了重金属钯和锡,同时偶联反应的杂质也相对较复杂。
专利cn104530044公布了如下的合成路线。
相较于之前的工艺路线,在制备化合物22时,该工艺使用甲苯作为溶剂,收率得到了大幅度的提升,同时也使得后处理变得简单。此外该工艺改变了酰胺化与甲基化的顺序,使用先酰化在还原的方式得到甲基,最后再与氯甲酸甲酯发生酰胺化制备利奥西呱。
专利cn105294686公布了如下的合成路线。
该工艺路线的特点在于更换了成环试剂,使得整个工艺避开了氢化反应,大大增加了工艺的安全性,但由于环合反应温度较高,化合物27结构不稳定。另外,该工艺以碳酸二甲酯作为酰胺化试剂,避免了氯甲酸甲酯带来的毒性。
专利cn1048924591公布了如下的合成路线。
该工艺路线别出心裁的以一步成环反应直接引入了甲基与甲酸甲酯基。彻底避免了氢化反应,但闭环反应在碱性条件下发生,而n-甲基-n-甲酸甲酯丙二腈单体在碱性体系下会不可避免的发生部分分解,同时,分解后的产物可能会继续与母体反应产生的杂质因为结构相似不易除去。
专利cn105367567、cn105367568和cn105367569先后公布了以下三个合成路线。
路线1与路线2以取代的方式避免了氢化反应,其设计思路基本类似,但对应的小分子单体难以商业化获得,小分子中间体的制备及纯化工艺难度较大。路线3中以苄基作为可离去基团,但仍然需要氢化还原反应。
纵观国内外各大工艺路线,各有特点,主要可分为两大类:一、不需要还原反应,这一类工艺普遍收率偏低,并且有难以除去的杂质;二、需要还原反应,这一类工艺多数都已ni/h2还原法制备关键中间体,但工艺化生产存在安全隐患。
技术实现要素:
本发明的目的是为了克服目前各工艺的不足之处,在现有技术以及总结经验的基础上进行了大量研究和探索,提供一种利奥西呱及其中间体制备方法。本发明收率高,操作简单安全,更适合大规模工业化生产。
本发明的目的可以通过以下措施达到:
一种利奥西呱的制备方法,其包括如下步骤:
(a)中间体la-1的制备:1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-甲脒盐酸盐与苯基偶氮丙二腈和碱在溶剂中反应,得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-[(e)-苯基二氮烯基]-4,6-嘧啶二胺;所述溶剂为甲基异丁基酮或二噁烷;
(b)中间体la-2的制备:2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-[(e)-苯基二氮烯基]-4,6-嘧啶二胺在还原剂作用下进行还原反应,得到2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺;所述还原剂选自水合肼、水合肼/催化剂;
(c)中间体la-3的制备:2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-4,5,6-嘧啶三胺与氯甲酸甲酯反应,得到4,6-二氨基-2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基氨基甲酸甲酯异丙醇溶剂合物;
(d)利奥西呱la的制备:4,6-二氨基-2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基氨基甲酸甲酯异丙醇溶剂合物在碱作用下与碘甲烷发生取代反应,得到4,6-二氨基-2-[1-(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基(甲基)氨基甲酸甲酯;
步骤(a)中,la-a与la-b在碱的作用下发生环合反应生成la-1。在现有技术,该步骤常以dmf作为反应溶剂,实验发现这会导致本步反应收率偏低。实验发现,该步骤可采用的反应溶剂包括异丁醇、正丁醇、丙醇、异丙醇、甲基异丁基酮、甲基乙基酮、二噁烷等,但只有甲基异丁基酮或二噁烷,特别是采用二噁烷溶剂时,可以大幅提高反应收率和产品纯度。本发明的该步骤避免了原研工艺的操作繁琐、收率低等缺点,使得本步工艺的收率和纯度比原专利提高,收率90%以上,比现有技术提高25%左右,并将纯度提至99%以上,且后处理简便、易于工业化生产。
步骤(a)中的碱为醇钠,优选甲醇钠或乙醇钠;该步骤的反应温度为60~120℃,优选90~103℃;反应物la-a:la-b:碱的摩尔比为1.0:1.0~1.5:2.0~2.5,优选1.0:1.0:2.0;反应时间为1~5h,优选1~2h。
步骤(a)中的反应结束后反应液于冰浴下搅拌析晶,最终可得到大量la-1。反应液通过直接抽滤,得到产品,操作简单,适合工艺化生产。
一种步骤(a)的具体反应如下:
步骤(b)中,la-1在单一水合肼或水合肼/催化剂的作用下发生还原反应生成la-2。现有技术中通常以pd/h2、ni/h2为还原试剂制备la-2,但在工业生产中pd/c、雷尼镍与h2的同时使用将会带来的巨大安全隐患,此外本步反应也需要特殊的氢化装置才能完成。实验发现该步骤中可选用的还原剂包括fe粉/h+、硼烷四氢呋喃络合物、硼氢化钠、氢化锂铝、连二亚硫酸钠、水合肼/催化剂、ni/环己烯、pd/环己烯等。
而本发明采用的水合肼或水合肼/催化剂为还原试剂,除了能够提高并稳定收率外,它还具有以下几个特点:采用水合肼或水合肼/催化剂体系作为还原剂,能够避免采用钯炭/h2还原体系易燃易爆、成本较高、反应周期较长、杂质较多等缺点,使得本步的工艺操作安全、反应周期短、操作可控、成本低廉;同时降低了生产设备的投入,更易于产业化。
步骤(b)中的水合肼/催化剂中的催化剂选自fe系催化剂、pd系催化剂、pt系催化剂、ni系催化剂、al3+/ni2+/ti4+复合催化剂中的一种或多种,优选ni或fe系催化剂。
在步骤(b)中的反应物la-1:ni或fe系催化剂:水合肼的摩尔比为1.0:0.1~3.0:5.0~10.0,优选1.0:0.5~1.0:6.0~8.0。该步骤的反应温度为20℃~80℃,优选25℃~60℃;反应时间为1~3h,优选1.5~2h。
一种步骤(b)的具体反应如下:
在步骤(c)中,la-2在异丙醇中与氯甲酸甲酯发生取代反应,经中和后得la-3;其具体步骤包括:la-2在异丙醇中与氯甲酸甲酯进行反应,然后加入三乙胺进行中和反应,反应后降至-5~5℃下进行结晶,得到化合物la-3。发明人发现,异丙醇对la-3溶解度较大,将la-3从异丙醇溶剂中直接抽滤得到的方法会使得本步反应收率偏低。本发明在后处理过程中将反应液置于冰浴下析晶,抽滤得la-3,收率明显上升。
步骤(c)中的反应物la-3:氯甲酸甲酯:三乙胺的摩尔比为1.0:1.2~1.4:1.4~1.6,优选1.0:1.3:1.5;la-2与氯甲酸甲酯的反应的温度为15~50℃,优选25~45℃;中和温度为15~70℃,优选45~55℃;反应时间为12~48h,优选20~22h。
一种步骤(c)的具体反应如下:
在步骤(d)中,la-3在强碱作用下与碘甲烷发生取代反应生成la粗品。在现有技术中,该步骤常以nah或双(三甲基甲硅烷基)氨基钠作为碱制备la,但在工业化生产中,nah/双(三甲基甲硅烷基)氨基钠在运输、使用、储存过程中极易吸水引发燃烧甚至爆炸。实验发现,该步骤中可采用的碱选自叔丁醇钾、叔丁醇钠、氢氧化铯、氢氧化钾或氢氧化钠等,发明人发现当以叔丁醇钾为碱并在其他条件的配合下,不仅可以稳定高收率,并能在保证纯度的前下提进一步提升安全性。
步骤(d)中化合物la-3与碱的摩尔比为1.0:1.8~2.5;该步骤中的反应温度为-5~5℃;。反应时间为2~6h,优选3~5h。
一种步骤(d)的具体反应如下:
本步发明采用的叔丁醇钾、叔丁醇钠、氢氧化铯、氢氧化钾或氢氧化钠等碱性试剂,除了能够提高并稳定收率外,它还具有以下几个特点:避免使用的的易燃易爆的钠氢、成本较高、杂质较多等缺点,使得本步的工艺操作简单、安全、成本低廉、更适合工业化生产。
综合以上工艺的描述,与现有技术相比,本发明具有如下优点:
1)la-1制备过程中以二噁烷为溶剂,避免了现有工艺的操作繁琐、收率低等缺点,使得本步工艺的收率和纯度比原专利提高,收率90%以上,比现有技术提高25%左右,并将纯度提至99%以上,且后处理简便、易于工业化生产;
2)la-2制备过程中以单一水合肼或水合肼/催化剂为还原试剂,避开了现有技术中使用的钯炭/h2还原体系易燃易爆、对生产设备的要求高、成本较高、反应周期较长、杂质较多等缺点,使得本步的工艺操作安全、反应周期短、清洁、成本低廉,节省了时间成本与人工成本;同时本发明的步骤反应温度低,降低了能耗,节约了成本;此外,本方法降低了生产设备的投入,更易于产业化;
3)la产品的制备过程中以叔丁醇钾、叔丁醇钠、氢氧化铯、氢氧化钾或氢氧化钠等碱性试剂代替原研工艺中使用的高活性碱性促进剂nah,避开了钠氢易燃易爆的缺点,提高了生产操作的安全性;同时提高并稳定了收率、降低了成本、减少了副产物的产生,使得本步的工艺操作简单、安全、成本低廉、更适合工业化生产。
4)本工艺还具有绿色环保的特点,本工艺中四步反应均收率高,选择性高,反应条件温和易于控制、固废少、能耗低,特别适合工业化生产。
附图说明
图1是本发明实施例6所得la粗品的1h-nmr;
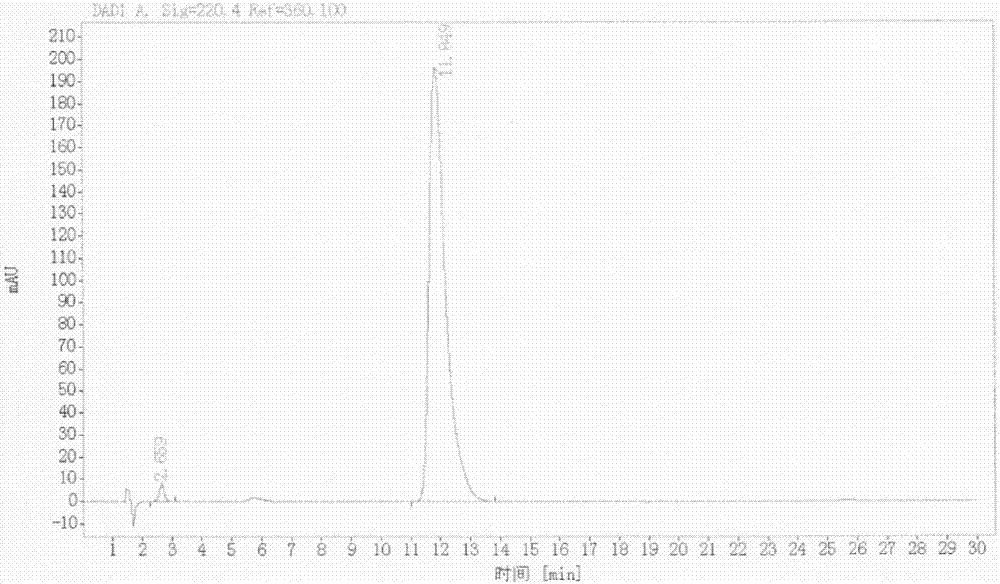
图2是本发明实施例6所得la粗品的高效液相色谱图;
图3是本发明实施例8所得la精制品的xrd。
具体实施方式
实施例1:la-1制备
向500ml四口瓶中加入二噁烷200ml,于搅拌下依次加入la-a(200.0g,0.65mol,1.0eq),la-b(110.0g,0.65mol,1.0eq),甲醇钠(70.0g,1.3mol,2.0eq),升温至100℃左右,缓慢回流,搅拌下反应1.0h。反应结束后,反应液搅拌下逐渐冷却至室温,于冰浴下继续搅拌2.0h。将反应液抽滤。抽滤,滤饼于50℃下干燥12~16h。得砖红色固体la-1:27.97g(收率97.4%,液相纯度99.53%,熔点>250℃)
1h-nmr(dmso-d6):δ=5.850(s,2h),7.136~7.166(m,1h),7.183~7.257(m,2h),7.345~7.418(m,3h),7.478~7.509(t,2h),7.856(s,2h)8.003~8.019(d,2h),8.418(s,2h)8.468~8.660(dd,1h),9.192~9.211(dd,1h)
对比例1:cn1330649c中page28实施例7a中描述如下:
将3.87g甲醇钠,然后将12.2g(71.7mmol)苯偶氮基丙二氰加入到进行着搅拌的21.92g(71.7mmol)得自实施例6a的1-(2-氟苄基)-1h-吡啶-3-甲脒在n,n-二甲基甲酰胺中的溶液中。将该混合物在110℃下搅拌一整夜,然后使之冷却。通过抽滤将沉淀出来的固体滤出,用乙醇对其进行洗涤,干燥后得到23g(理论值的73%)目标化合物。
缺点:使用了n,n-二甲基甲酰胺溶剂,转化率较低,产品的收率仅为73%。
对比例2:cn104530044c中page8实施例1中描述如下:
步骤一,在2l三口瓶中,依次加入1-(2-氟苄基)-1h-[3,4-b]吡啶-3-甲脒盐酸盐(化合物1,94.5g,0.31mol)、1.2l甲苯,搅拌下依次加入甲醇钠(17g,0.31mol),苯基偶氮丙二氰(化合物2,252.5g,0.31mol),加热至120℃回流,tlc检测反应完毕,冷却至室温,抽滤,滤饼用甲苯洗一次,再用水打浆固体,抽滤,烘干的到中间体化合物3(129g,0.29mol,收率95%,含量99%)。
缺点:使用了甲苯溶剂,反应温度较本工艺所采用的1,4-二氧六环高约10℃,相对能耗高。
通过对其制备工艺的多次重复实验,发现其反应时间为5h以上,且其工艺中存在一个较大的工艺杂质(由苯基偶氮丙二氰引入),难以除去,对后续步骤(la-2的制备)有重大影响,造成后面步骤反应时间延长,且副反应较多。而本发明采用二氧六环作为反应溶剂,可以降低由苯基偶氮丙二氰引入的杂质的风险,样品中几乎没有此杂质,对后续的反应(la-2的制备)没有影响。
重要的是利用本工艺的反应时间较短,仅需反应1~2h,即可反应完全,使得反应周期缩短,节约了成本。
此外,利用本工艺制备的产品收率和纯度均优于cn104530044c中所制备的样品,收率高达97.4%、hplc纯度达99.53%。
实施例2:la-1制备
向100ml四口瓶中加入甲基异丙基酮60ml,于搅拌下依次加入la-a(6.0g,0.020mol,1.0eq),la-b(3.3g,0.020mol,1.0eq),甲醇钠(2.1g,0.039mol,2.0eq),升温至100℃,搅拌下反应2.0h。反应结束后,反应液搅拌下逐渐冷却至室温,于冰浴下继续搅拌2.0h。将反应液抽滤,滤饼以60ml水中悬浮洗涤。抽滤,滤饼于50℃下干燥16h。得砖红色la-1:7.3g(收率85.1%,液相纯度99.13%,熔点>250℃)
实施例3:la-2制备
于100ml四口瓶中加入dmf(30ml),搅拌下依次加入la-1(5.0g,0.012mol,1.0eq),活性炭(0.5g),fecl3·6h2o(0.8g,0.003mol,0.25eq)。搅拌下升温至60℃,缓慢滴加80%水合肼(15.0g,0.238mol,10.0eq)。滴加完毕后于60℃下反应20~24h。反应结束后,抽滤,除去活性炭,以25mldmf洗涤滤饼,所得滤液于85℃下旋蒸除去溶剂。向旋蒸后的浓缩物中加入25ml甲醇,于冰浴下滴加25ml水,滴加完毕后于冰浴下继续搅拌1h,抽滤,滤饼于50℃下干燥16h,得土黄色la-2:3.4g(收率:86.4%,液相纯度98.1%,熔点244.4~245.8℃)
1h-nmr(dmso-d6):4.068(s,2h),5.782(s,2h),5.836(s,4h),7.101~7.165(m,2h),7.200~7.237(m,1h),7.280~7.356(m,2h),8.576~8.585(m,1h),9.055~9.071(d,1h)
实施例4:la-2制备
于500ml四口瓶中加入dmf(200ml),搅拌下依次加入la-1(20.0g,0.048mol,1.0eq),雷尼ni(1.3g,0.023mol,0.5eq)。搅拌下升温至30℃,缓慢滴加80%水合肼(23.0g,0.368mol,8.0eq)。滴加完毕后于30℃下反应1.0h。反应结束后,抽滤,除去雷尼ni,以50mldmf洗涤滤饼,所得滤液于85℃下旋蒸除去溶剂。向旋蒸后的浓缩物中加入100ml甲醇,于冰浴下滴加100ml水,滴加完毕后于冰浴下继续搅拌2h,抽滤,滤饼于50℃下干燥16h,得土黄色la-2:14.1g(收率:88.2%,液相纯度98.1%,熔点244.4~245.8℃)对比例:cn1330649c中page29实施例8a中描述如下:
将5g(11.38mmol)得自实施例7a的2-[1-(2-氟苄基)-1h)-吡唑并[3,4-b]吡啶-3-基]-5-[(e)-苯基二氮烯基]-4,6-嘧啶二胺用800mg50%阮内镍水溶液在60mldmf中在65巴的氢气压下在62℃氢化22小时。通过用硅藻土通过抽滤进行过滤滤出催化剂,将溶液在真空下蒸发,然后用5n盐酸进行搅拌。用抽滤将分离出来的淡黄褐色沉淀滤出并干燥,得到3.1g(理论值的59.3%)目标化合物。通过与稀碳酸氢钠溶液一起进行振摇并用乙酸乙酯进行萃取得到游离碱。用抽滤将在两相中不溶的固体滤出。乙酸乙酯相还包少量的游离碱。
缺点:使用了易燃易爆的雷尼镍(阮内镍)和氢气,危险系数较高,需要特殊的氢化装置,操作性差,不便于工业化生产;且反应时间太长,副反应增多;此外,产品后处理程序复杂,收率较低。
实施例5:la-3制备
于500ml四口瓶中加入100ml异丙醇,搅拌下加入la-2(10.0g,0.0285mol,1.0eq),升温至35℃,搅拌30min后缓慢滴加氯甲酸甲酯(14.0g,0.148,1.3eq),滴加完毕于35℃下反应16h。反应结束后,将反应液升温至50℃,搅拌下缓慢滴加三乙胺(7.6g,0.171mol,1.5eq)。滴加完毕后于50℃下继续搅拌2h。反应结束后将反应液自然冷却至室温,于冰浴下继续搅拌2.0h。抽滤,滤饼于50℃下真空干燥16h,得黄色固体la-3:10.1g(收率80.8%,液相纯度99.13%,熔点206.1~208.0℃)
1h-nmr(dmso-d6):3.630(s,3h),5.812(s,2h),6.163(s,4h),7.134(s,2h),7.203~7.239(m,1h),7.337~7.346(d,2h),8.007(s,1h),8.605(s,1h),9.068~9.083(d,1h)
实施例6:la粗品制备
于250ml四口瓶中加入dmf(100ml),搅拌下加入la-3(5.0g,0.0114mol,1.0eq),搅拌5min,反应液不能溶清。反应液在冰盐浴下降温至~5~0℃,搅拌下加入叔丁醇钾(2.1g,0.0183mol,1.6eq),加料完毕后继续于~5~0℃下搅拌2.0~3.0h。于~5~0℃下滴加碘甲烷(2.9g,0.021mol,1.8eq),滴加过程中控制温度不超过0℃,滴加完毕后保温继续反应3.5~4.5h。反应结束后加入氢氧化钾溶液(2mol/l)猝灭。以dcm分别萃取反应液两次,合并有机层以无水硫酸钠干燥,抽滤,滤液以旋蒸浓缩至油状物。向浓缩物中加入50ml水,于室温下打浆2h,抽滤,滤饼于50℃下真空干燥16h,得淡黄色固体la粗品4.2g(收率87.2%,纯度99.5%,熔点>250℃)
1h-nmr(dmso-d6):3.023(s,3h),3.545~3.668(m,3h),5.805(s,2h),6.331~6.351(d,4h),7.109~7.118(d,2h),7.199~7.236(s,1h),7.313~7.364(s,2h),8.595~8.604(s,1h),9.053~9.071(dd,1h)
对比例:cn1330649c中page35实施例6中描述如下:
将54mg(0.13mmol)得自实施例3的4,6-二氨基-2-[1(2-氟苄基)-1h-吡唑并[3,4-b]吡啶-3-基]-5-嘧啶基氨基甲酸乙酯溶于5mldmf中,将该混合物冷却至0℃,向其中加入7.67mg(0.19mmol)氢化钠。然后向其中滴加18.14mg(0.13mmol)碘代甲烷,然后将该混合物搅拌一小时。将该混合物与水混合并用二氯甲烷进行萃取。将所合并的有机相用硫酸镁干燥,用旋转蒸发器浓缩。将残余物首先用柱色谱进行纯化,然后用制备rp-hplc进行纯化。收率:32mg(理论值的58%)。
缺点:使用了易燃易爆的氢化钠,危险系数较高,操作性差,不便于工业化生产;且产品后处理程序复杂,成本较高,但收率较低。
实施例7:la粗品制备
于1000ml四口瓶中加入dmf(400ml),搅拌下加入la-3(20.0g,0.046mol,1.0eq),搅拌5min,反应液不能溶清。反应液在冰盐浴下降温至~5~0℃,搅拌下加入叔丁醇钾(10.3g,0.092mol,2.0eq),加料完毕后继续于~5~0℃下搅拌2.0h。于-5~0℃下滴加碘甲烷(13.1g,0.092mol,2.0eq),滴加过程中控制温度不超过0℃,滴加完毕后保温继续反应5.0h。反应结束后加入2mol/l氢氧化钾溶液(70ml)猝灭。以200mldcm分别萃取反应液两次,合并有机层以无水硫酸钠干燥,抽滤,滤液以旋蒸浓缩至油状物。向浓缩物中加入200ml水,于室温下打浆2h,抽滤,滤饼于50℃下真空干燥16h,得淡黄色固体la粗品1.68g(收率87.2%,纯度99.3%,熔点>250℃)
实施例8:la重结晶
将如实施例10所制得的la粗品6g溶于15mldmso与7.5ml乙酸乙酯中,加热回流下(85℃)固体溶清,加入1g活性炭,以7.5ml乙酸乙酯冲洗,回流下搅拌10min。抽滤,以7.5ml乙酸乙酯洗涤滤饼,滤液于回流下缓慢加入80ml乙酸乙酯,回流下继续搅拌30min。除去加热,自然冷却至室温并继续搅拌6h。过滤,将所得白色固体置于90ml乙酸乙酯中回流下搅拌1h,冷却至室温,继续搅拌2h。过滤,所得固体于50℃下干燥16h,得白色固体:4.7g(收率78.3%,纯度99.9%,熔点>250℃)
上述内容以具体实施例的形式对本发明进行了进一步阐述。需要注意的是,本发明并不局限于上述特定实施案例。
- 还没有人留言评论。精彩留言会获得点赞!