咔唑衍生物,以及使用咔唑衍生物的发光元件、发光器件和电子器件的制作方法
本申请是以下申请的分案申请:申请日:2008年11月28日;申请号:200880126449.5(pct/jp2008/072104,201410545381.1);发明名称:“咔唑衍生物,以及使用咔唑衍生物的发光元件、发光器件和电子器件”的分案申请。
本发明涉及咔唑衍生物、使用咔唑衍生物的发光元件、发光器件和电子器件。
背景技术:
近年来,对于用电致发光的发光元件的研究和开发非常活跃。作为这些发光元件的基本结构,将含发光物质的层插入电极对之间。通过向该元件施加电压,可由发光物质得到光发射。
因为这种发光元件是自发光类型,它具有比液晶显示元件更优的优点,例如高可见度的像素且不需要背光源(backlight),因此认为适用于平板显示元件。另外,可将这种发光元件制成薄和重量轻的元件,这也是重要优点。另外,极高反应速度也为其特征。
另外,因为可将这种发光元件形成膜形式,可通过形成大面积元件,容易的得到平面光发射。难以通过使用由白炽灯或led代表的点光源或通过使用由荧光灯代表的线光源得到该特性。因此,上述发光元件还具有作为适用于照明等的平面光源的高实用性价值。
根据发光物质是有机化合物还是无机化合物,将此类用电致发光的发光元件粗略分类。当有机化合物用于发光物质时,通过向发光元件施加电压,然后电流通过其中,将电极对中的电子和空穴注入含发光有机化合物的层。然后通过将这些载体(电子和空穴)重组,发光有机化合物形成激发态,当激发态返回基态时则发射光。
因为这种机理,这种发光元件称为电流激励发光元件。注意,有机化合物的激发态可为单重激发态或三重激发态。由单重激发态发射的光称为荧光,由三重激发态发射的光称为磷光。
在改善这种发光元件的元件特性中,存在取决于物质的很多问题,为解决这些问题,已进行了元件结构改善、物质开发等方面的工作(例如非专利文件1:meng-huanho,yao-shanwuandchinh.chen,2005sidinternationalsymposiumdigestoftechnicalpapers,第xxxvi卷.第802-805页)。
在非专利文件1中所述发光元件中,4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)用作与发光层接触的层。但是,npb具有低的单态激发能,且存在能量可从处于激发态的发光材料转移的可能性。因为在发射具有短波长的蓝光的发光材料的情况中,激发态的能量水平特别高,能量转移到npb的可能性更高。因为能量转移到npb,存在发光元件的发光效率降低的问题。
技术实现要素:
因此。本发明的目的是通过提供新的咔唑衍生物提供具有高发光效率的发光元件。另外,本发明的另一个目的是提供能耗低并在低电压下驱动的发光器件和电子器件。
本发明的一个特征是由以下通式(1)代表的咔唑衍生物
在该式中,α1、α2、α3和α4各自代表具有小于或等于13个碳原子的亚芳基,这些碳原子形成环;ar1和ar2各自代表具有小于或等于13个碳原子的芳基,这些碳原子形成环;r1代表氢原子、具有1-6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团;r2代表具有1-6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团。另外,l、m和n各自独立为0或1。
另外,在以上结构中,通式(1)中α1-α4由以下通式(2-1)-(2-12)中任一个式代表。
在式中,r11–r16、r21-r30、r31-r38和r41-r45各自代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。r46和r47各自代表具有1–6个碳原子的烷基和苯基中的任何基团。另外,r46和r47可相互连接形成环。r48代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。
另外,在以上结构中,通式(1)中ar1和ar2由以下通式(3-1)-(3-6)中任一个式代表。
在式中,r51-r56、r61-r70、r71-r78和r81-r85各自代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。r86和r87各自代表具有1–6个碳原子的烷基和苯基中的任何基团。另外,r86和r87可相互连接形成环。r88和r89各自代表氢原子、具有1-6个碳原子的烷基、苯基和联苯基中的任何基团。
另外,在以上结构中,通式(1)中r1由以下通式(4-1)-(4-9)任一个式代表,通式(1)中r2由以下通式(4-2)-(4-9)任一个式代表。
在式中,r51-r70各自代表氢原子、具有1-6个碳原子的烷基、苯基和联苯基中的任何基团。
另外,本发明的一个特征由以下结构式(5)-(8)中任一个式代表。
另外,作为本发明的另一个特征,发光元件包括电极对之间的el层,el层至少包括发光层和空穴-传输层,发光层和空穴-传输层中至少一个含上述任何咔唑衍生物。
另外,作为本发明的另一个特征,发光元件包括位于阳极和阴极之间的el层,el层至少包括发光层、空穴-传输层和空穴-注入层,形成的空穴-注入层与阳极接触,发光层、空穴-传输层和空穴-注入层中至少一个含上述任何咔唑衍生物。
另外,在以上结构中,可使用结构,其中空穴-注入层含上述任何咔唑衍生物和相对于咔唑衍生物显示电子接收性质的无机化合物。注意,可使用过渡金属的氧化物作为无机化合物。另外,作为无机化合物,可使用氧化钛、氧化钒、氧化钼、氧化钨、氧化铼、氧化钌、氧化铬、氧化锆、氧化铪、氧化钽和氧化银中的一种或多种。
另外,作为本发明的另一个特征,用任何上述发光元件形成发光器件,用该发光器件形成电子器件。
另外,本发明还包括具有上述发光元件的发光器件和具有该发光器件的电子器件(devuce)。本说明书中的发光器件是指图像显示装置、发光器件或光源(包括照明装置)。另外,发光器件包括以下所有模块:其中连接器例如柔性印刷电路(fpc)、带式自动连接(tab)带,或带式运输器包装(tcp)与发光器件连接的模块;在tab带或tcp的末端提供有印刷线路板的模块;和其中通过玻璃上芯片技术(cog)方法将集成电路(ic)直接安装在发光元件上的模块。
因为本发明咔唑衍生物显示高空穴-传输性质,所以它可主要用于空穴-传输层,该空穴-传输层包含在发光元件的el层中。另外,本发明咔唑衍生物用于形成发光元件的空穴-传输层,由此可形成具有高发光效率的发光元件。
另外,可用该发光元件得到发光器件和电子器件,它们能耗低且可在低电压下驱动。
附图简述
在附图中:
图1a和1b为截面图,各自显示实施方案方式2中的发光元件的叠层结构;
图2a-2c为截面图,各自显示实施方案方式2中的发光元件的发光方式;
图3为截面图,显示实施方案方式3中的发光元件的叠层结构;
图4a和4b分别为实施方案方式4中有源矩阵发光器件的顶视图和截面图;
图5a和5b分别为实施方案方式4中无源矩阵发光器件的透视图和截面图;
图6a-6d是各自显示实施方案方式5中电子器件的视图;
图7为显示用本发明发光器件作为背光源的液晶显示装置的视图;
图8为显示使用本发明发光器件的台灯的视图;
图9为显示使用本发明发光器件的室内照明装置的视图;
图10a和10b为显示pcba1bp(缩写)的1hnmr图谱的图;
图11a和11b为显示pcba1bp(缩写)的吸收光谱和发射光谱的图;
图12a和12b为显示pcbbi1bp(缩写)的1hnmr图谱的图;
图13a和13b为显示pcbbi1bp(缩写)的吸收光谱和发射光谱的图;
图14a和14b为显示pcbaf(缩写)的1hnmr图谱的图;
图15a和15b为显示pcbaf(缩写)的吸收光谱和发射光谱的图;
图16a和16b为显示pcbasf(缩写)的1hnmr图谱的图;
图17a和17b为显示pcbasf(缩写)的吸收光谱和发射光谱的图;
图18为显示实施方案5中发光元件的元件结构的截面图;
图19为显示发光元件1和发光元件2的电流密度与亮度特性之间的关系图;
图20为显示发光元件1和发光元件2的电压与亮度特性之间的关系图;
图21为显示发光元件1和发光元件2的亮度与电流效率特性之间的关系图;
图22为显示发光元件1和发光元件2的电压与电流特性的关系图;
图23为显示发光元件1和发光元件2的发射光谱的图;
图24为显示通过恒定电流驱动进行的发光元件1和发光元件2的连续照明试验的结果图;
图25为显示发光元件1和发光元件3的电流密度与亮度特性之间的关系图;
图26为显示发光元件1和发光元件3的电压与亮度特性之间的关系图;
图27为显示发光元件1和发光元件3的亮度与电流效率特性之间的关系图;
图28为显示发光元件1和发光元件3的电压与电流特性之间的关系图;
图29为显示发光元件1和发光元件3的发射光谱的图;
图30为显示发光元件1和发光元件4的电流密度与亮度特性之间的关系图;
图31为显示发光元件1和发光元件4的电压与亮度特性之间的关系图;
图32为显示发光元件1和发光元件4的亮度与电流效率特性的关系图;
图33为显示发光元件1和发光元件4的电压与电流特性之间的关系图;
图34为显示发光元件1和发光元件4的发射光谱的图;
图35为显示通过恒定电流驱动进行的发光元件1和发光元件4的连续照明试验的结果图;
图36为显示发光元件1和发光元件5的电流密度与亮度特性之间的关系图;
图37为显示发光元件1和发光元件5的电压与亮度特性之间的关系图;
图38为显示发光元件1和发光元件5的亮度与电流效率特性之间的关系图;
图39为显示发光元件1和发光元件5的电压与电流特性之间的关系图;
图40为显示发光元件1和发光元件5的发射光谱的图;
图41为显示pcba1bp(缩写)的cv特性的图;
图42为显示pcbbi1bp(缩写)的cv特性的图;
图43为显示pcbaf(缩写)的cv特性的图;
图44为显示pcbasf(缩写)的cv特性的图;
图45a和45b为显示pcta1bp(缩写)的1hnmr图谱的图;
图46a和46b为显示pctbi1bp(缩写)的1hnmr图谱的图;
图47a和47b为显示pcbanb(缩写)的1hnmr图谱的图;
图48a和48b为显示pcbnbb(缩写)的1hnmr图谱的图;
图49a和49b为显示pcbbinb(缩写)的1hnmr图谱的图;
图50a和50b为显示pcbant(缩写)的1hnmr图谱的图;
图51a和51b为显示bcba1bp(缩写)的1hnmr图谱的图;
图52a和52b为显示bcbanb(缩写)的1hnmr图谱的图;
图53a和53b为显示bcbbinb(缩写)的1hnmr图谱的图;
图54a和54b为显示nbcbalbp(缩写)的1hnmr图谱的图;
图55a和55b为显示ncba1bp(缩写)的1hnmr图谱的图;
图56为显示发光元件1和发光元件6–8的电压与亮度特性的图;
图57为显示发光元件1和发光元件6–8的亮度与电流效率特性之间的关系图;
图58为显示发光元件1和发光元件6–8的电压与电流特性之间的关系图;
图59为显示发光元件1和发光元件6–8的发射光谱的图;
图60为显示通过恒定电流驱动进行的发光元件1和发光元件6-8的连续照明试验的结果图;
图61a和61b为显示pcbbi1bpiii(缩写)的1hnmr图谱的图;
图62a-62c为显示pcba1bpiv(缩写)的1hnmr图谱的图;
图63a和63b为显示pcbnbbβ(缩写)的1hnmr图谱的图;和
图64a和64b为显示pcbbiflp(缩写)的1hnmr图谱的图。
实施本发明的最佳方式
以下通过参考附图,详细的描述本发明的实施方案方式和实施方案。但是,本发明不限于以下给出的描述,可容易的理解,在不偏离本发明目的和范围的前提下,可对方式及其细节进行各种修改。因此,本发明不应视为限于以下实施方案方式和实施方案的描述。
[实施方案方式1]
在实施方案方式1中,将描述本发明咔唑衍生物。
本发明咔唑衍生物由通式(1)代表。
在式中,α1、α2、α3和α4各自代表具有小于或等于13个碳原子的亚芳基,所述碳原子形成环;ar1和ar2各自代表具有小于或等于13个碳原子的芳基,所述碳原子形成环;r1代表氢原子、具有1–6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团;r2代表具有1–6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团。另外,l、m和n各自独立为0或1。
在通式(1)中,α1-α4各自代表具有小于或等于13个碳原子的亚芳基,所述碳原子形成环。尤其是,可给出由结构式(2-1)-(2-12)代表的取代基。
在式中,r11–r16、r21-r30、r31-r38和r41-r45各自代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。r46和r47各自代表具有1–6个碳原子的烷基和苯基中的任何基团。另外,r46和r47可相互连接,形成环。r48代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。
在通式(1)中,ar1和ar2各自代表具有小于或等于13个碳原子的芳基,所述碳原子形成环。尤其是,可给出由结构式(3-1)-(3-6)代表的取代基。
在式中,r51-r56、r61-r70、r71-r78和r81–r85各自代表氢原子、具有1-6个碳原子的烷基、苯基和联苯基中的任何基团。r86和r87各自代表具有1–6个碳原子的烷基和苯基中的任何基团。另外,r86和r87可相互连接形成环。r88和r89各自代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。
在通式(1)中,r1代表氢原子、具有1-6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团;r2代表具有1-6个碳原子的烷基、取代或未取代的苯基,和取代或未取代的联苯基中的任何基团。尤其是,可为r1给出由结构式(4-1)-(4-9)代表的取代基,可为r2给出由结构式(4-2)-(4-9)代表的取代基。
在式中,r51-r70各自代表氢原子、具有1–6个碳原子的烷基、苯基和联苯基中的任何基团。
作为由通式(1)代表的本发明咔唑衍生物的具体实例,可给出由结构式(9)-(425)代表的咔唑衍生物。但是,本发明不限于此。
另外,可通过由以下合成流程(a-1)-(a-7)、合成流程(b-1)和合成流程(c-1)-(c-2)代表的合成方法合成由通式(1)代表的本发明咔唑衍生物。
[卤代仲芳胺(化合物a)的合成方法]
可按如以下合成流程(a-1)的方法合成由通式代表的卤代仲芳胺(化合物a)。换言之,首先,通过用卤化剂将仲芳胺(化合物a1)卤化,可由此得到卤代仲芳胺(化合物a)。注意,作为卤化剂,可使用n-溴代琥珀酰亚胺(nbs)、n-碘代琥珀酰亚胺(nis)、溴、碘、碘化钾等。另外,各x1代表卤基,优选为溴或碘。
[卤代咔唑衍生物(化合物b2)的合成方法]
可按如以下合成流程(a-2)的方法合成由通式代表的卤代咔唑衍生物(化合物b2)。换言之,首先,用卤化剂将咔唑衍生物(化合物b1)卤化,由此可得到卤代咔唑衍生物(化合物b2)。注意,作为卤化剂,可使用n-溴代琥珀酰亚胺(nbs)、n-碘代琥珀酰亚胺(nis)、溴、碘、碘化钾等。另外,各x1代表卤基,优选为溴或碘。
[其中9h-咔唑-3-硼酸或9h-咔唑的3位被有机硼取代的化合物(化合物b)的合成方法]
可按如以下合成流程(a-3)的方法合成由通式代表的其中9h-咔唑的3位被硼酸或有机硼取代的化合物(化合物b)。换言之,在卤代咔唑衍生物(化合物b2)上,用烷基锂试剂和硼试剂进行硼氧化或有机硼化,由此可得到其中9h-咔唑的3位被硼酸或有机硼取代的化合物(化合物b)。
注意,流程(a-3)中r99代表具有1–6个碳原子的烷基。r98代表具有1-6个碳原子的烷基。另外,r100和r101各自代表氢原子或具有1-6个碳原子的烷基。r102和r103可相互连接形成环。另外,正丁基锂、甲基锂等可作为烷基锂试剂使用。硼酸三甲酯、硼酸异丙酯等可作为硼试剂使用。
[仲芳胺(化合物c3)的合成方法]
可按如以下合成流程(a-4)的方法合成由通式代表的仲芳胺(化合物c3)。换言之,在碱的存在下,用金属催化剂使卤代芳基(化合物c1)与伯芳胺(化合物c2)偶合,由此可得到仲芳胺(化合物c3)。
在进行buchwald-hartwig反应的情况下,尽管作为可用于合成流程(a-4)中的钯催化剂,可给出二(二亚苄基丙酮)合钯(0)、乙酸钯(ii)等,但可使用的钯催化剂不限于此。尽管作为可用于合成流程(a-4)的钯催化剂的配体,可给出三(叔丁基)膦、三(正己基)膦、三环己基膦等,但可使用的配体不限于此。
尽管作为可用于合成流程(a-4)的碱,可给出有机碱例如叔丁醇钠、无机碱例如碳酸钾等,但可使用的碱不限于此。另外,尽管作为可用于合成流程(a-4)的溶剂,可给出甲苯、二甲苯、苯、四氢呋喃等,但可使用的溶剂不限于此。
描述了在合成流程(a-4)中进行ullmann反应的情况。在合成流程(a-4)中,r104和r105各自代表卤基、乙酰基等,可给出氯、溴和碘作为卤基。优选r104为碘,形成碘化亚铜(i),或r105为乙酰基,形成乙酸铜(ii)。用于反应的铜化合物不限于此,可用铜作为铜化合物的替代。尽管作为可用于合成流程(a-4)的碱,可给出无机碱例如碳酸钾,但可使用的碱不限于此。
尽管作为可用于合成流程(a-4)的溶剂,可给出1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu)、甲苯、二甲苯、苯等,但可使用的溶剂不限于此。优选使用具有高沸点的dmpu或二甲苯,因为通过ullmann反应,当反应温度大于或等于100℃时,可在较短时间内达到目的并得到较高收率。因为还优选反应温度大于或等于150℃,所以更优选使用dmpu。
[叔芳胺(化合物c5)的合成方法]
可按如以下合成流程(a-5)的方法合成由通式代表的叔芳胺(化合物c5)。换言之,在碱的存在下,用金属催化剂使仲芳胺(化合物c3)和卤代芳基(化合物c4)偶合,由此可得到叔芳胺(化合物c5)。
在进行buchwald-hartwig反应的情况下,尽管作为可用于合成流程(a-5)的钯催化剂,可给出二(二亚苄基丙酮)合钯(0)、乙酸钯(ii)等,但可使用的钯催化剂不限于此。尽管作为可用于合成流程(a-5)的钯催化剂的配体,可给出三(叔丁基)膦、三(正己基)膦、三环己基膦等,但可使用的配体不限于此。
尽管作为可用于合成流程(a-5)的碱,可给出有机碱例如叔丁醇钠、无机碱例如碳酸钾等,可使用的碱不限于此。另外,尽管作为可用于合成流程(a-5)的溶剂,可给出甲苯、二甲苯、苯、四氢呋喃等,但可使用的溶剂不限于此。
描述在合成流程(a-5)中进行ullmann反应的情况。在合成流程(a-5)中,r104和r105各自代表卤基、乙酰基等,可给出氯、溴和碘作为卤基。优选r104为碘,形成碘化亚铜(i),或r105为乙酰基,形成乙酸铜(ii)。用于反应的铜化合物不限于此,铜可用作铜化合物的备选。尽管作为可用于合成流程(a-5)的碱,可给出无机碱例如碳酸钾,但可使用的碱不限于此。
尽管作为可用于合成流程(a-5)的溶剂,可给出1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu)、甲苯、二甲苯、苯等,但可使用的溶剂不限于此。优选使用具有高沸点的dmpu或二甲苯,因为通过ullmann反应,可在较短时间内达到目的,且当反应温度大于或等于100℃时收率较高。因为还优选反应温度大于或等于150℃,所以更优选使用dmpu。
[叔芳胺(化合物c5)的合成方法]
可按如以下合成流程(a-6)的方法合成由通式代表的叔芳胺(化合物c5)。换言之,在碱的存在下,用金属催化剂使伯芳胺(化合物c2)和卤代芳基(化合物c1和c4)偶合,由此可得到叔芳胺(化合物c5)。但是,当ar1和ar2相同、β1和β2相同、l和m相同时,可得到化合物c5,收率高。
在进行buchwald-hartwig反应的情况下,尽管作为可用于合成流程(a-6)的钯催化剂,可给出二(二亚苄基丙酮)合钯(0)、乙酸钯(ii)等,但可使用的钯催化剂不限于此。尽管作为可用于合成流程(a-6)的钯催化剂的配体,可给出三(叔丁基)膦、三(正己基)膦、三环己基膦等,但可使用的配体不限于此。
尽管作为可用于合成流程(a-6)的碱,可给出有机碱例如叔丁醇钠、无机碱例如碳酸钾等,但可使用的碱不限于此。另外,尽管作为可用于合成流程(a-6)的溶剂,可给出甲苯、二甲苯、苯、四氢呋喃等,但可使用的溶剂不限于此。
描述在合成流程(a-6)中进行ullmann反应的情况。在合成流程(a-6)中,r104和r105各自代表卤基、乙酰基等,可给出氯、溴和碘作为卤基。优选r104为碘,形成碘化亚铜(i),或r105为乙酰基,形成乙酸铜(ii)。用于反应的铜化合物不限于此,铜可用作铜化合物的备选。尽管作为可用于合成流程(a-6)的碱,可给出无机碱例如碳酸钾,但可使用的碱不限于此。
尽管作为可用于合成流程(a-6)的溶剂,可给出1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu)、甲苯、二甲苯、苯等,但可使用的溶剂不限于此。优选使用具有高沸点的dmpu或二甲苯,因为通过ullmann反应,可在较短时间内达到目的,且当反应温度大于或等于100℃时收率较高。因为还优选反应温度大于或等于150℃,所以更优选使用dmpu。
[卤代叔芳胺衍生物(化合物c)的合成方法]
可按如以下合成流程(a-7)的方法合成由通式代表的卤代叔芳胺(化合物c)。换言之,首先,用卤化剂将叔芳胺(化合物c5)卤化,由此可得到卤代叔芳胺(化合物c)。注意,可用n-溴代琥珀酰亚胺(nbs)、n-碘代琥珀酰亚胺(nis)、溴、碘、碘化钾等作为卤化剂。另外,各x1代表卤基,优选为溴或碘。
[仲芳胺(化合物d)的合成方法]
可按如以下合成流程(b-1)的方法合成由通式代表的具有咔唑的仲芳胺(化合物d)。换言之,可在碱的存在下,用金属催化剂使卤代仲芳胺(化合物a)和其中9h-咔唑的3位被硼酸或有机硼取代的化合物(化合物b)偶合。从而可得到具有咔唑的仲芳胺(化合物d)。
在以上任一流程中,描述了使用suzuki-miyaura反应的情况。作为可用作金属催化剂的钯催化剂,可给出乙酸钯(ii)、四(三苯基膦)合钯(0)、二(三苯基膦)二氯合钯(ii)等。作为以上钯催化剂的配体,可给出三(邻-甲苯基)膦、三苯基膦、三环己基膦等。另外,作为以上碱,可给出有机碱例如叔丁醇钠、无机碱例如碳酸钾等。作为可使用的溶剂,可给出甲苯和水的混合溶剂;甲苯、醇例如乙醇和水的混合溶剂;二甲苯和水的混合溶剂;二甲苯、醇例如乙醇和水的混合溶剂;苯和水的混合溶剂;苯、醇例如乙醇和水的混合溶剂;醚例如乙二醇二甲醚和水的混合溶剂等。
但是,可使用的催化剂、配体、碱和溶剂不限于此。
另外,在以上任一流程中,除芳基硼酸外,使用有机铝、有机锆、有机锌或有机锡化合物等的交叉偶联产物可用作基础物质(basematerial)。但是,本发明不限于此。
[具有咔唑的叔芳胺(化合物e)的合成方法]
可按如以下合成流程(c-1)的方法合成由通式代表的具有咔唑的叔芳胺(化合物e)。换言之,可在碱的存在下,用金属催化剂使具有咔唑的芳胺(化合物d)和卤代芳基(化合物c4)偶合,由此可得到具有咔唑的叔芳胺(化合物e),该化合物为最终产物。
在以上任一流程中,描述了使用suzuki-miyaura反应的情况。作为可用作金属催化剂的钯催化剂,可给出乙酸钯(ii)、四(三苯基膦)合钯(0)、二(三苯基膦)二氯合钯(ii)等。作为以上钯催化剂的配体,可给出三(邻-甲苯基)膦、三苯基膦、三环己基膦等。另外,作为以上碱,可给出有机碱例如叔丁醇钠、无机碱例如碳酸钾等。作为可使用的溶剂,可给出甲苯和水的混合溶剂;甲苯、醇例如乙醇和水的混合溶剂;二甲苯和水的混合溶剂;二甲苯、醇例如乙醇和水的混合溶剂;苯和水的混合溶剂;苯、醇例如乙醇和水的混合溶剂;醚例如乙二醇二甲醚和水的混合溶剂等。
但是,可使用的催化剂、配体、碱和溶剂不限于此。
另外,在以上任一流程中,除芳基硼酸外,使用有机铝、有机锆、有机锌、有机锆、有机锡等的交叉偶联产物可用作基础物质。但是,本发明不限于此。
[具有咔唑的叔芳胺(化合物e)的另一种合成方法]
可按如以下合成流程(c-2)的方法合成由通式代表的具有咔唑的叔芳胺(化合物e)。换言之,首先,可在碱的存在下,用金属催化剂使卤代叔芳胺(化合物c)和其中9h-咔唑的3位被硼酸或有机硼取代的化合物(化合物b)偶合,由此可得到具有咔唑的叔芳胺(化合物e),该化合物为最终产物。
[实施方案方式2]
在实施方案方式2中,描述用实施方案方式1中所述本发明咔唑衍生物形成的发光元件,用于空穴-传输层。
实施方案方式2中的发光元件包括起阳极作用的第一电极、起阴极作用的第二电极,和插入第一电极和第二电极之间的el层。注意,当将电压施加到各电极以便第一电极的电位高于第二电极时,实施方案方式2中的发光元件可得到光发射。
另外,实施方案方式2中的发光元件的el层在其结构中包括第一层(空穴-注入层)、第二层(空穴-传输层)、第三层(发光层)、第四层(电子-传输层)和第五层(电子-注入层),位于第一电极一侧。
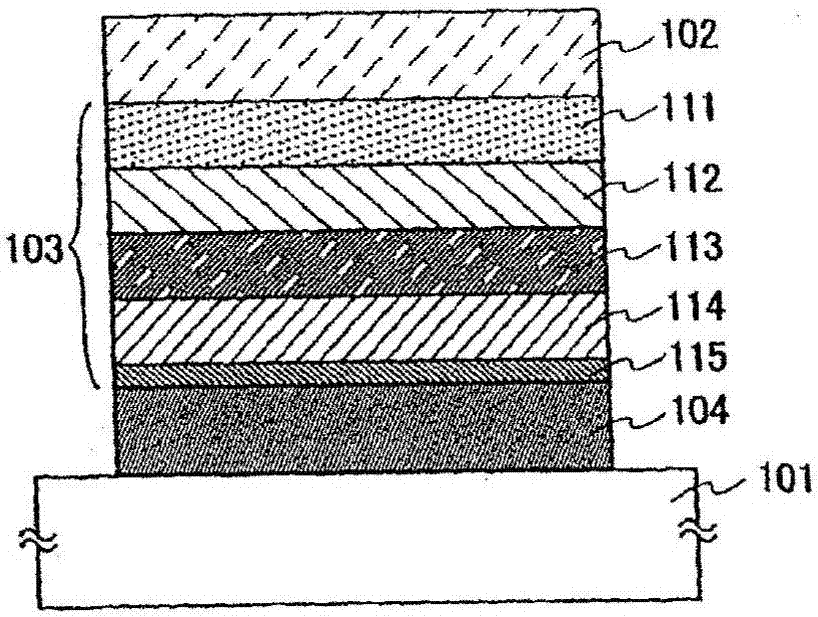
参照图1a和1b描述实施方案方式2中发光元件的结构。基底101用作发光元件的载体。对于基底101,例如可使用玻璃、石英、塑料等。
注意,尽管以上基底101可保留在使用本发明发光元件的发光器件或电子器件中,但在发光元件的制备方法中,基底101仅可具有发光元件的载体功能,不在最终产品中保留。
对于在基底101上形成的第一电极102,优选使用具有高功函(尤其是,4.0ev或更高功函)的金属、合金、导电化合物、其混合物等。尤其是,可给出以下实例:铟锡氧化物(ito)、含硅或氧化硅的铟锡氧化物、铟锌氧化物(izo),和含氧化钨和氧化锌的氧化铟。另外,还可给出金(au)、铂(pt)、镍(ni)、钨(w)、铬(cr)、钼(mo)、铁(fe)、钴(co)、铜(cu)、钯(pd)、钛(ti)、金属材料的氮化物(例如氮化钛)等。但是,在本发明中,用复合材料形成el层103中的第一层111,其形成后与第一电极102接触,无论第一电极102的功函大小,可容易的向复合材料注入空穴。因此,可使用各种已知方法,只要其是可用作电极材料的材料(例如也包括金属、合金、导电化合物、其混合物等,或属于元素周期表中的第1或2族的元素)。
通常通过溅射法形成那些材料中任一材料的膜。例如,可通过溅射法,用靶(target)形成铟锌氧化物(izo),其中将1%(重量)-20%(重量)氧化锌加入氧化铟;且可通过溅射法,用靶形成含氧化钨和氧化锌的氧化铟,其中将0.5%(重量)-5%(重量)氧化钨和0.1%(重量)-1%(重量)氧化锌加入氧化铟。或者,可通过真空蒸发法、喷墨法、旋涂法等形成第一层111。
另外,当含在下文中描述的复合材料的层作为材料用于形成的第一层111时,第一层111与el层103中的第一电极102接触,el层103在第一电极102之上形成,任何各种材料例如金属、合金和导电化合物;其混合物等可作为用于第一电极102的物质,与它们的功函无关。例如,也可使用铝(al)、银(ag)、含铝的合金(alsi)等。
另外,也可使用属于元素周期表第1或2族的元素,它们为低功函材料,即碱金属例如锂(li)或铯(cs);碱土金属例如镁(mg)、钙(ca)或锶(sr)、含这些金属中任何金属的合金(例如mgag合金或alli合金);稀土金属例如铕(eu)或镱(yb);含此类稀土金属的合金等。
注意,在其中用碱金属、碱土金属或其合金形成第一电极102的情况下,可使用真空蒸发法或溅射法。注意,在使用银糊等的情况下,可使用涂层法、喷墨法等。
对于在第一电极102上形成的el层103,可使用已知物质,可使用任何低分子量化合物和大分子化合物。注意,用于形成el层103的物质不仅具有仅由有机化合物形成的结构,而且还具有部分含无机化合物的结构。
为形成el层103,使含具有高空穴-注入性质物质的空穴-注入层、含具有高空穴-传输性质物质的空穴-传输层、含发光物质的发光层、含具有高电子-传输性质物质的电子-传输层、含具有高电子-注入性质物质的电子-注入层等相互组合,并在合适时层叠。
注意,在图1a所示el层103中,将在第一电极102一侧的第1层(空穴-注入层)111、第2层(空穴-传输层)112、第3层(发光层)113、第4层(电子-传输层)114和第5层(电子-注入层)115按顺序层叠。
作为空穴-注入层的第一层111为含具有高空穴-注入性质材料的空穴-注入层。作为具有高空穴-注入性质的材料,可使用氧化钼、氧化钛、氧化钒、氧化铼、氧化钌、氧化铬、氧化锆、氧化铪、氧化钽、氧化银、氧化钨、氧化锰等。或者,作为低分子量有机化合物,可给出酞菁-基化合物例如酞菁(缩写:h2pc)、酞菁铜(ii)(缩写:cupc)或钒氧酞菁(vanadylphthalocyanine)(缩写:vopc)。
另外,还可给出以下为低分子量有机化合物的芳胺化合物:
1)4,4',4"-三(n,n-二苯基氨基)三苯胺(缩写:tdata);
2)4,4',4"-三[n-(3-甲基苯基)-n-苯基氨基]三苯胺(缩写:mtdata);
3)4,4'-二[n-(4-二苯基氨基苯基)-n-苯基氨基]联苯(缩写:dpab);
4)4,4'-二(n-{4-[n'-(3-甲基苯基)-n'-苯基氨基]苯基}-n-苯基氨基)联苯(缩写:dntpd);
5)1,3,5-三[n-(4-二苯基氨基苯基)-n-苯基氨基]苯(缩写:dpa3b);
6)3-[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(缩写:pczpca1);
7)3,6-二[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(缩写:pczpca2);
8)3-[n-(1-萘基)-n-(9-苯基咔唑-3-基)氨基]-9-苯基咔唑(缩写:pczpcn1)等。
注意,也可按类似方式使用实施方案方式1中所述本发明咔唑衍生物。
另外,还可使用大分子化合物(低聚物、树枝状大分子、聚合物等)。例如,可给出大分子化合物例如
1)聚(n-乙烯基咔唑)(缩写:pvk);
2)聚(4-乙烯基三苯胺)(缩写:pvtpa),
3)聚[n-(4-{n’-[4-(4-二苯基氨基)苯基]苯基-n’-苯基氨基}苯基)异丁烯酰胺](缩写:ptpdma),和
4)聚[n,n’–二(4-丁基苯基)-n,n’–二(苯基)联苯胺](缩写:poly-tpd)。另外,也可使用向其中加入酸的大分子化合物,例如聚(3,4-乙二氧基噻吩)/聚(苯乙烯磺酸)(pedot/pss)或聚苯胺/聚(苯乙烯磺酸)(缩写:pani/pss)。
或者,对于第一层111,可使用复合材料,其中具有受体性质的材料包含在具有高空穴-传输性质的材料中。注意,通过使用具有高空穴-传输性质的含具有受体性质的物质的物质,无论其功函大小,可选择用于形成电极的材料。换言之,不但具有高功函的材料而且具有低功函的材料也可用作第一电极102。可通过使具有高空穴-传输性质的物质与具有受体性质的物质共蒸发,形成此类复合材料。注意,在本说明书中,“组合物”表示在材料之间给予和接受电荷的环境下不仅是两种材料的简单混合物,而且也是许多材料的混合物。
可用各种化合物例如芳胺化合物、咔唑衍生物、芳烃和大分子化合物(低聚物、树枝状大分子、聚合物等)作为用于复合材料的有机化合物。用于复合材料的有机化合物优选为具有高空穴-传输性质的有机化合物。尤其是,优选使用具有空穴迁移率10-6cm2/vs或更高迁移率的物质。但是,可使用不同于以上的物质,只要该物质的空穴-传输性质大于电子-传输性质。以下具体显示可用于复合材料的有机化合物。
可给出用于复合材料的有机化合物,例如芳胺化合物例如mtdata、tdata、dpab、dntpd、dpa3b、pczpcal、pczpca2、pczpcn1、4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb或α-npd),和n,n’–二(3-甲基苯基)-n,n’-二苯基-[1,1'-联苯]-4,4'-二胺(缩写:tpd);和咔唑衍生物例如4,4'-二(n-咔唑基)联苯(缩写:cbp)、1,3,5-三[4-(n-咔唑基)苯基]苯(缩写:tcpb)、9-[4-(n-咔唑基)]苯基-10-苯基蒽(缩写:czpa),和1,4-二[4-(n-咔唑基)苯基]-2,3,5,6-四苯基苯。注意,也可按类似方式使用在实施方案方式1中描述的本发明咔唑衍生物。
另外,可给出以下芳烃化合物:
1)2-叔丁基-9,10-二(2-萘基)蒽(缩写:t-budna);
2)2-叔丁基-9,10-二(1-萘基)蒽;
3)9,10-双(3,5-二苯基苯基)蒽(缩写:dppa);
4)2-叔丁基-9,10-双(4-苯基苯基)蒽(缩写:t-budba);
5)9,10-二(2-萘基)蒽(缩写:dna);
6)9,10-二苯基蒽(缩写:dpanth);
7)2-叔丁基蒽(缩写:t-buanth);
8)9,10-二(4-甲基-1-萘基)蒽(缩写:dmna);
9)9,10-二[2-(1-萘基)苯基]-2-叔丁基-蒽;
10)9,10-二[2-(1-萘基)苯基]蒽;
11)2,3,6,7-四甲基-9,10-二(1-萘基)蒽等。
另外,也可给出以下芳烃化合物:
1)2,3,6,7-四甲基-9,10-二(2-萘基)蒽;
2)9,9'-联蒽;
3)10,10'-二苯基-9,9'-联蒽;
4)10,10'-二(2-苯基苯基)-9,9'-联蒽;
5)10,10'-二[(2,3,4,5,6-五苯基)苯基]-9,9'-联蒽;
6)蒽;
7)并四苯;
8)红荧烯;
9)苝;
10)2,5,8,11-四(叔丁基)苝;
11)并五苯;
12)蒄;
13)4,4'-二(2,2-二苯基乙烯基)联苯(缩写:dpvbi);
14)9,10-二[4-(2,2-二苯基乙烯基)苯基]蒽(缩写:dpvpa)等。
可给出有机化合物例如7,7,8,8-四氰基-2,3,5,6-四氟醌二甲烷(缩写:f4-tcnq)和四氯对醌,及过渡金属氧化物作为具有受体性质的物质。另外,可给出属于元素周期表的第4–8族的金属的氧化物。尤其是,因为高电子接收性质,优选氧化钒、氧化铌、氧化钽、氧化铬、氧化钼、氧化钨、氧化锰和氧化铼。其中,因为其在空气中稳定及其吸湿性质低,以致于其可容易的处理,所以尤其优选氧化钼。
注意,用以上大分子化合物例如pvk、pvtpa、ptpdma或聚tpd和具有受体性质的以上物质形成的复合材料可用于第一层111。注意,通过使实施方案方式1中描述的本发明咔唑衍生物与具有受体性质的以上物质组合形成的复合材料也可用于第一层111。
作为空穴-传输层的第二层112为含具有高空穴-传输性质的物质的空穴-传输层。注意,在实施方案方式1中描述的本发明咔唑衍生物用于实施方案方式2中第二层112。
另外,在实施方案方式1中描述的本发明咔唑衍生物也可用于第一层111和第二层112。在该情况下,可容易的制造元件,也可提高材料使用效率。另外,因为第一层111和第二层112的能量图相同或相似,可在第一层111和第二层112之间容易的运输载体。
第三层113为发光层,该发光层含具有高发光性质的物质。对于第三层113,可使用以下给出的任何低分子量有机化合物。
作为用于发射蓝光的发光物质,可给出n,n'-二[4-(9h-咔唑-9-基)苯基]-n,n’-二苯基均二苯乙烯-4,4'-二胺(缩写:yga2s)、4-(9h-咔唑-9-基)-4'-(10-苯基-9-蒽基)三苯胺(缩写:ygapa)等。
作为发射绿光的发光物质,可给出以下:
1)n-(9,10-二苯基-2-蒽基)-n,9-二苯基-9h-咔唑-3-胺(缩写:2pcapa);
2)n-[9,10-二(1,1'-联苯-2-基)-2-蒽基]-n,9-二苯基-9h-咔唑-3-胺(缩写:2pcabpha);
3)n-(9,10-二苯基-2-蒽基)-n,n',n'-三苯基-1,4-苯二胺(缩写:2dpapa);
4)n-[9,10-二(1,1'-联苯-2-基)-2-蒽基]-n,n',n'-三苯基-1,4-苯二胺(缩写:2dpabpha);
5)n-[9,10-二(1,1'-联苯-2-基)]-n-[4-(9h-咔唑-9-基)苯基]-n-苯基蒽-2-胺(缩写:2ygabpha);
6)n,n,9-三苯基蒽-9-胺(缩写:dphapha)等。
作为发射黄光的发光物质,可给出红荧烯、5,12-二(1,1'-联苯-4-基)-6,11-二苯基并四苯(缩写:bpt)等。另外,作为发射红光的发光物质,可给出n,n,n',n'-四(4-甲基苯基)并四苯-5,11-二胺(缩写:p-mphtd)、7,13-二苯基-n,n,n',n'-四(4-甲基苯基)苊并[1,2-a]荧蒽-3,10-二胺(缩写:p-mphafd)等。
另外,第三层113可具有其中具有高发光性质的以上物质分散在另一种物质中的结构。注意,在分散的情况下,待分散的物质浓度优选设为总量的20%(质量)或更小。另外,可使用已知物质,作为其中具有发光性质的物质被分散的物质。优选使用以下物质:该物质的最低未占分子轨道水平(lumo水平)比具有发光性质的物质的深(绝对值较大),且最高占据分子轨道水平(homo水平)比具有发光性质的物质的浅(绝对值较小)。
尤其是,可使用以下任何金属络合物:
1)三(8-羟基喹啉根(quinolinolato))合铝(iii)(缩写:alq);
2)三(4-甲基-8-羟基喹啉根)合铝(iii)(缩写:almq3);
3)双(10-羟基苯并[h]喹啉酸根(quinolinato))合铍(ii)(缩写:bebq2);
4)双(2-甲基-8-羟基喹啉根)(4-苯基苯酚根(phenolato))合铝(iii)(缩写:ba1q);
5)双(8-羟基喹啉根)合锌(ii)(缩写:znq);
6)双[2-(2-苯并
7)双[2-(2-苯并噻唑基)苯酚根]合锌(ii)(缩写:znbtz)等。
另外,可使用以下任何杂环化合物:
1)2-(联苯-4-基)-5-(4-叔丁基苯基)-1,3,4-
2)1,3-二[5-(对-叔丁基苯基)-1,3,4-
3)3-(联苯-4-基)-4-苯基-5-(4-叔丁基苯基)-1,2,4-三唑(缩写:taz);
4)2,2',2"-(1,3,5-苯三苯甲基)三(1-苯基-1h-苯并咪唑)(缩写:tpbi);
5)红菲绕啉(缩写:bphen);
6)浴铜灵(bcp)等。
另外,也可使用以下任何稠合芳族化合物:
9-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(缩写:czpa);
3,6-二苯基-9-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(缩写:dpczpa);
9,10-二(3,5-二苯基苯基)蒽(缩写:dppa);
9,10-二(2-萘基)蒽(缩写:dna);
2-叔丁基-9,10-二(2-萘基)蒽(缩写:t-budna);
9,9'-联蒽(缩写:bant);
9,9'-(均二苯乙烯-3,3'-二基)联菲(缩写:dpns);
9,9'-(均二苯乙烯-4,4'-二基)联菲(缩写:dpns2);
3,3',3"-(苯-1,3,5-三苯甲基)三芘(缩写:tpb3)等。
可用多种物质作为其中具有发光性质的物质被分散的物质。例如,为抑制结晶,还可加入用于抑制红荧烯等结晶的物质。另外,还可加入npb、alq等,以有效的将能量转移至具有发光性质的物质。因此,由于存在其中具有高发光性质的物质分散在另一种物质的结构,可抑制第三层113结晶。另外,可抑制由高浓度的具有高发光性质的物质产生的浓度淬灭。
另外,在以上物质中,尤其优选使用具有电子传输性质的物质,以使具有发光性质的物质分散在其中,形成第三层113。尤其是,也可使用以上任何金属络合物和杂环化合物;以上稠合芳族化合物中的czpa、dna和t-budna;和下文中给出的其它大分子化合物,作为可用于第四层114的物质。
或者,对于第三层113,可使用以下大分子化合物。
作为发射蓝光的发光物质,可给出:
聚(9,9-二辛基芴-2,7-二基)(缩写:pof),
[(9,9-二辛基芴-2,7-二基)-(2,5-二甲氧基苯-1,4-二基)]共聚物(缩写:pf-dmop),
{(9,9-二辛基芴-2,7-二基)-[n,n’-二(对-丁基苯基)-1,4-二氨基苯]}共聚物(缩写:tab-pfh)等。
作为发射绿光的发光物质,可给出:
聚(对-亚苯基亚乙烯基)(缩写:ppv),
[(9,9-二己基芴-2,7-二基)-(苯并[2,1,3]噻二唑-4,7-二基)]交替共聚物(缩写:pfbt),
[(9,9-二辛基-2,7-二亚乙烯基亚芴基(fluorenylene))-(2-甲氧基-5-(2-乙基己氧基)-1,4-亚苯基)]交替共聚物等。
作为发射橙色-红色光的发光物质,可给出:
聚[2-甲氧基-5-(2'-乙基己氧基)-1,4-亚苯基亚乙烯基](缩写:meh-ppv),
聚(3-丁基噻吩-2,5-二基)(缩写:r4-pat),
{[9,9-二己基-2,7-二(1-氰基亚乙烯基)亚芴基]-[2,5-二(n,n’–二苯基氨基)-1,4-亚苯基]}交替共聚物,
{[2-甲氧基-5-(2-乙基己氧基)-1,4-二(1-氰基亚乙烯基亚苯基)]-[2,5-二(n,n’–二苯基氨基)-1,4-亚苯基]}交替共聚物(缩写:cn-ppv-dpd)等。
第四层114为电子-传输层,该层含具有高电子-传输性质的物质。对于第四层114,例如可使用作为低分子量有机化合物的金属络合物,例如alq、almq3、bebq2、balq、znq、znpbo或znbtz等。或者,可使用杂环化合物例如pbd、oxd-7、taz、tpbi、bphen或bcp代替金属络合物。本文中所述物质主要为具有10-6cm2/vs或更高电子迁移率的物质。注意,除以上物质外的其它物质可用于电子-传输层,只要该物质的电子-传输性质大于其空穴-传输性质。另外,电子-传输层不限于单层,而是也可为由以上物质形成的两层或更多层的叠层。
或者,对于第四层114,可使用大分子化合物。例如,可使用[(9,9-二己基芴-2,7-二基)-(吡啶-3,5-二基)]共聚物(缩写:pf-py)、[(9,9-二辛基芴-2,7-二基)-(2,2'-联吡啶-6,6'-二基)]共聚物(缩写:pf-bpy)等。
另外,第五层115为电子-注入层,该层含具有高电子-注入性质的物质。对于第五层115,可使用碱金属、碱土金属或其化合物例如氟化锂(lif)、氟化铯(csf)或氟化钙(caf2)。或者,可使用由具有电子-传输性质的物质形成的层,其含碱金属、碱土金属或其化合物,尤其是,由含镁(mg)等的alq形成的层。注意,在该情况下,电子可从第二电极104更有效地注入。
对于第二电极104,可使用具有低功函(尤其是,3.8ev或更低功函)的金属、合金、导电化合物、其混合物等。作为这种阴极材料的具体实例,可给出属于元素周期表中第1或2族的元素即碱金属例如锂(li)或铯(cs);碱土金属例如镁(mg)、钙(ca)或锶(sr);含这些金属中任何金属的合金(例如mgag合金或alli合金);稀土金属例如铕(eu)或镱(yb);含此类稀土金属的合金等。
注意,在用碱金属、碱土金属及其合金形成第二电极104的情况下,可使用真空蒸发法或溅射法。注意,在用银糊等的情况下,可使用涂层法、喷墨法等。
注意,通过提供第五层115,可用任何种类的导电材料例如al、ag、ito和含硅或氧化硅的铟锡氧化物形成第二电极104,无论它们的功函大小。可通过溅射法、喷墨法、旋涂法等形成这些导电材料。
另外,作为el层103的形成方法,其中将第一层(空穴-注入层)111、第二层(空穴-传输层)112、第三层(发光层)113、第四层(电子-传输层)114和第五层(电子-注入层)115按顺序堆叠,可使用各种方法中的任何方法,无论方法为干法或湿法。例如,可使用真空蒸发法、喷墨法、旋涂法等。注意,对于各层,可使用不同形成方法。
除干法例如溅射法或真空蒸发法外,也可通过湿法例如溶胶-凝胶法,用金属材料糊形成第二电极104。
在上述本发明发光元件中,由于第一电极102和第二电极104之间形成的电位差导致电流流动,以及空穴和电子在el层103中重组,从而发射光。然后,通过第一电极102和第二电极104中之一或二者,该发射光在外部被提取(extracted)。因此,第一电极102和第二电极104中之一或二者为具有透光性质的电极。
注意,当仅第一电极102为具有透光性质的电极时,如图2a所示,通过第一电极102,从基底101一侧提取由el层103发射的光。或者,当仅第二电极104为具有透光性质的电极时,如图2b所示,通过第二电极104,从基底101一侧的对面提取由el层103发射的光。再或者,当第一电极102和第二电极104均为具有透光性质的电极时,如图2c所示,通过第一电极102和第二电极104,将el层103发射的光提取到基底101一侧和基底101一侧的对面。
在第一电极102和第二电极104之间提供层的结构不限于以上。可使用除以上以外的结构,只要至少包括第二层112空穴-传输层和第三层113发光层。
或者,如图1b所示,可使用结构,其中起阴极作用的第二电极104、el层103和起阳极作用的第一电极102按顺序堆叠在基底101上。注意,在该情况下,el层103具有结构,其中第五层115、第四层114、第三层113、第二层112、第一层111和第一电极102按顺序堆叠在第二电极104上。
注意,通过使用本发明发光元件,可制备其中发光元件的驱动(drive)受薄膜晶体管(tft)控制的无源矩阵发光器件或有源矩阵发光器件。
注意,在制备有源矩阵发光器件的情况下,对tft的结构无特别限制。例如,适当时可使用交错tft或反向交错tft。另外,可由n型tft和p型tft或仅由n型tft或p型tft形成在tft基底上形成的激励电路。另外,对用于tft的半导体膜的结晶度无特别限制。无定形半导体膜或结晶半导体膜均可用于tft。
因为在发光元件中用本发明咔唑衍生物形成第二层(空穴-传输层)112,在实施方案方式2中显示该发光元件,所以可实现不仅提高元件效率,而且还抑制驱动电压增加。
注意,适当时,可使实施方案方式2与实施方案方式1中所述任何结构组合。
[实施方案方式3]
在实施方案方式3中,参考附图3,描述发光元件,该发光元件具有实施方案方式2中所述任何发光元件的许多el层(下文中称为堆叠型发光元件)。该发光元件为在第一电极301和第二电极302之间具有许多el层(第一el层303和第二el层304)的堆叠型发光元件。注意,尽管在实施方案方式3中描述二el层结构,但也可使用三或更多el层结构。
在实施方案方式3中,第一电极301起阳极作用,第二电极302起阴极作用。注意,对于第一电极301和第二电极302,可使用类似于实施方案方式1中所述那些的结构。另外,对于许多el层(第一el层303和第二el层304),可使用类似于实施方案方式2中所述那些的结构。注意,第一el层303和第二el层304的结构彼此可相同或不同,且可与实施方案方式2中所述那些相似。
另外,在许多el层(第一el层303和第二el层304)之间提供电荷发生层305。当将电压施加到第一电极301和第二电极302时,电荷发生层305具有将电子注入el层之一和将空穴注入其他el层的功能。在实施方案方式3中,当施加电压使第一电极301的电位高于第二电极302时,电荷发生层305将电子注入第一el层303和将空穴注入第二el层304。
注意,优选电荷发生层305具有在光提取效率方面的透光性质。另外,即使当其电导率小于第一电极301或第二电极302时,电荷发生层305也起作用。
电荷发生层305可具有结构,其中将具有受体性质的物质加入具有高空穴传输性质的物质;或结构,其中将具有供体性质的物质加入具有高电子传输性质的物质。或者,可将这两种结构叠加在一起。
在使用其中将具有受体性质的物质加入具有高空穴传输性质的物质的结构的情况下,作为具有高空穴传输性质的物质,可使用例如芳胺化合物例如4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb或α-npd)、n,n’-二(3-甲基苯基)-n,n’–二苯基-[1,1'-联苯]-4,4'-二胺(缩写:tpd)、4,4',4"-三(n,n–二苯基氨基)三苯胺(缩写:tdata)、4,4',4"-三[n-(3-甲基苯基)-n-苯基氨基]三苯胺(缩写:mtdata)或4,4'-二[n-(螺-9,9'-联芴-2-基)-n-苯基氨基]-1,1'-联苯(缩写:bspb)。此处所述物质主要为空穴迁移率大于或等于10-6cm2/vs的物质。注意,也可使用上述物质以外的物质,只要其空穴传输性质大于其电子传输性质。
另外,可给出7,7,8,8-四氰基-2,3,5,6-四氟醌二甲烷(缩写:f4-tcnq)、氯醌等作为具有受体性质的物质。另外,还可给出过渡金属氧化物。另外,可给出属于元素周期表中第4-8族的金属的氧化物。尤其是,因为具有高电子接受性质,优选氧化钒、氧化铌、氧化钽、氧化铬、氧化钼、氧化钨、氧化锰和氧化铼。其中,尤其优选氧化钼,因为它在空气中稳定及其低吸湿性质而易于处理。
在另一方面,在使用其中将具有供体性质的物质加入到具有高电子传输性质的物质的结构的情况下,作为具有高电子传输性质的物质,可使用例如具有喹啉骨架或苯并喹啉骨架的金属络合物,例如三(8-羟基喹啉根)合铝(iii)(缩写:alq)、三(4-甲基-8-羟基喹啉根)合铝(iii)(缩写:almq3)、二(10-羟基苯并[h]喹啉酸根)合铍(ii)(缩写:bebq2)或二(2-甲基-8-羟基喹啉根)(4-苯基苯酚根)合铝(iii)(缩写:balq)。另外,具有
另外,对于具有供体性质的物质,可使用碱金属、碱土金属、稀土金属、属于元素周期表中第13族的金属或其氧化物或碳酸盐。尤其优选使用锂(li)、铯(cs)、镁(mg)、钙(ca)、镱(yb)、铟(in)、氧化锂、碳酸铯等。或者,有机化合物例如四硫代并四苯(tetrathianaphthacene)可用作具有供体性质的物质。
注意,通过使用任何以上材料形成电荷发生层305,在el层堆叠的情况下,可抑制驱动电压增加。
尽管在实施方案方式3中描述了具有双el层的发光元件,但本发明可类似的适用于其中3或更多el层堆叠的发光元件。如实施方案方式3的发光元件,通过用位于电极对之间的电荷发生层使许多el层彼此分离排列,可在高亮度区域中得到长寿命元件,同时保持低电流密度。作为应用实例,在发光元件应用于照明的情况下,可减少因电极材料的电阻所致电压降。因此,光可在大区域范围内均匀发射。另外,可得到功耗低且在低电压下驱动的发光器件。
另外,当el层具有不同发射颜色时,可由全发光元件得到需要的发射颜色。例如,在具有双el层的发光元件中,当使第一el层的发射颜色和第二el层的发射颜色成为补色时,也可得到发射白光的发光元件,作为全发光元件。注意,“补色”表示颜色之间的关系,当它们混合时,颜色变为消色颜色。即可通过由发射补色光的物质得到的混合光得到白光发射。
在具有三el层的发光元件中,例如当第一el层的发射颜色为红色,第二el层的发射颜色为绿色,第三el层的发射颜色为蓝色时,作为全发光元件也可类似的得到白光。
注意,适当时,可将实施方案方式3与实施方案方式1和2中所述任何结构组合。
[实施方案方式4]
在实施方案方式4中,参照图4a和4b,描述在像素部分中具有本发明发光元件的发光器件。图4a为发光器件的顶视图,图4b为沿着图4a中a-a'和b-b'取的截面图。
在图4a中,虚线所示参考符号401、402和403分别表示激励电路部分(源激励电路)、像素部分和激励电路部分(门激励电路)。参考符号404和405分别表示封闭基底和密封剂,由密封剂405封闭的内侧区域为空间407。
引线408是将输入的信号传送至源激励电路部分401和门激励电路403,和从用作外输入端的柔性印刷电路(fpc)409接收视频信号、时钟信号、起动信号、调节信号等的线。尽管此处仅显示fpc,但该fpc可提供有印刷线路板(pwb)。另外,本说明书中的发光器件不仅包括发光器件本身,而且还包括与fpc或pwb附接的发光器件。
然后,参照图4b描述发光器件的截面结构。在元件基底410上形成激励电路部分和像素部分。在此显示像素部分402中一个像素和为激励电路部分的源激励电路401。形成通过使n-通道tft423和p-通道tft424组合得到的cmos电路,作为源激励电路401。可通过各种cmos电路、pmos电路或nmos电路形成激励电路。在实施方案方式4中,尽管描述的是其中在基底上形成激励电路的驱动器集成(driver-integrated)型结构,但激励电路不是必需在基底上形成,而可在基底以外形成。
像素部分402由许多像素形成,这些像素具有开关tft411、电流控制tft412和第一电极413,第一电极413与电流控制tft412的漏极(drain)电连接。形成绝缘体414,以覆盖第一电极413的末端部分。
优选形成绝缘体414,以便具有在其上末端部分或下末端部分弯曲的弯曲表面,以得到有利覆盖。例如,通过用正型光敏丙烯酸作为绝缘体414的材料,可形成具有弯曲表面的绝缘体414,该弯曲表面仅在上末端部分具有曲率半径(0.2μm-3μm)。另外,可使用在蚀刻剂中通过光照成为不溶性的负型光敏材料或在蚀刻剂中通过光照射成为可溶性的正型光敏材料,作为绝缘体414。
el层416和第二电极417形成在第一电极413之上。此处,可用各种材料中的任何材料例如金属、合金和导电化合物或其混合物形成第一电极413。注意,作为特定材料,可用实施方案方式2中所示材料作为可用于第一电极的材料。
另外,通过各种方法中的任何方法例如使用蒸发罩的蒸发法、喷墨法或旋涂法形成el层416。el层416具有实施方案方式2中所述结构。可使用低分子量化合物或大分子化合物(包括低聚物或树枝状大分子),作为包含在el层416中的另一种材料。也可用不仅有机化合物而且无机化合物,作为el层的材料。
作为第二电极417的材料,可使用各种金属、合金和导电化合物或其混合物的任何材料。在第二电极417用作阴极的情况下,其中优选使用低功函(3.8ev或更低功函)的金属、合金、导电化合物、其混合物等。例如,可给出属于元素周期表的第1或2族的元素,即碱金属例如锂(li)或铯(cs);碱土金属例如镁(mg)、钙(ca)或锶(sr)、或含这些金属中任何金属的合金(例如mgag合金或alli合金)等。
注意,在el层416中发生的光通过第二电极417传送的情况下,对于第二电极417,也可使用具有减少的厚度的金属薄膜和透明导电膜(铟锡氧化物(ito)、含硅或氧化硅的铟锡氧化物、铟锌氧化物(izo)或含氧化钨和氧化锌的氧化铟等)的叠层。
通过用密封剂405使密封基底404和元件基底410附接,得到结构,其中在被元件基底410、密封基底404和密封剂405包围的空间407中提供发光元件418。注意,空间407中充满填料例如惰性气体(例如氮气、氩气等)或密封剂405。
优选使用环氧基树脂作为密封剂405。另外,优选尽可能的不传递水分和氧气的材料。可使用由frp(纤维玻璃-增强塑料)、pvf(聚氟乙烯)、聚酯、丙烯酸等形成的塑料基底以及玻璃基底或石英基底,作为密封基底404的材料。
如上所述,可得到具有本发明发光元件的有源矩阵发光器件。
另外,除以上有源矩阵发光器件外,本发明发光元件还可用于无源矩阵发光器件。图5a和5b分别显示使用本发明发光元件的无源矩阵发光器件的透视图和截面图。注意,图5a为发光器件的透视图,图5b为图5a沿着线x-y取的截面图。
在图5a和5b中,在基底501之上的第一电极502和第二电极503之间提供el层504。将第一电极502的边缘部分用绝缘层505覆盖。然后,在绝缘层505之上提供隔离层506。隔离层506的侧壁具有斜面,以使当侧壁与基底表面接近时,一个侧面与另一侧面之间的距离变得更窄。换言之,在短边方向上的隔离层506的截面为梯形,下部基部(面朝与绝缘层505的平面方向相似的方向,并与绝缘层505接触的一侧)比上部基部(面朝与绝缘层505的平面方向相似的方向,但与绝缘层505不接触的一侧)短。通过按这种方式提供隔离层506,可避免因静电等导致的发光元件的缺陷。
通过以上方法,可得到使用本发明发光元件的无源矩阵发光器件。
注意,用具有高发光效率的本发明发光元件形成实施方案方式4中所述任何发光器件(有源矩阵发光器件和无源矩阵发光器件),从而得到具有减少的功耗的发光器件。
注意,适当时,可使实施方案方式4与实施方案方式1–3中所述任何结构组合。
[实施方案方式5]
在实施方案方式5中,描述电子器件,该电子器件作为其一部分包括实施方案方式4中所示本发明发光器件。该电子器件的实例包括照象机例如摄像机或数码照象机、眼镜型(goggle)显示器、导航系统、声音再现装置(例如汽车话音系统和音频组件)、计算机、游戏机、便携式信息终端(例如便携式计算机、移动电话、便携游戏机和电子书),其中提供记录介质的图像再现装置(尤其是,能够使记录介质再现的装置例如数字通用唱片(dvd)和配备可显示图像的显示器单元的装置)等。这些电子器件的具体实例在图6a-6d中显示。
图6a显示本发明电视机,该电视机包括外壳611、支承基座612、显示器部分613、扬声器部分614、视频输入端615等。在该电视机中,可将本发明发光器件施用于显示器部分613。因为本发明发光器件具有高发光效率的特征,可通过施用本发明发光器件得到具有减少的功耗的电视机。
图6b显示本发明计算机,该计算机包括主体621、外壳622、显示器部分623、键盘624、外部连接端口625、指点器626等。在该计算机中,可将本发明发光器件施用于显示器部分623。因为本发明发光器件具有高发光效率的特征,可通过施用本发明发光器件得到具有减少的功耗的计算机。
图6c显示本发明移动电话,该移动电话包括主体631、外壳632、显示器部分633、音频输入部分634、音频输出部分635、操作键636、外部连接端口637、天线638等。在该移动电话中,可将本发明发光器件施用到显示器部分633。因为本发明发光器件具有高发光效率的特征,可通过施用本发明发光器件得到具有减少的功耗的移动电话。
图6d显示本发明照相机,该照相机包括主体641、显示器部分642、外壳643、外部连接端口644、遥控接收部分645、图像接收部分646、电池647、声频输入部分648、操作键649、目镜部分650等。在该照相机中,可将本发明发光器件施用于显示器部分642。因为本发明发光器件具有高发光效率的特征,可通过施用本发明发光器件得到具有减少的功耗的照相机。
如上所述,本发明发光器件的适用范围如此宽,以致于该发光器件可应用于各种领域中的电子器件。
本发明发光器件可用作照明装置。图7为液晶显示装置的实例,其中本发明发光器件用作背光源。图7中显示的液晶显示装置包括外壳701、液晶层702、背光源703和外壳704。液晶层702与激励ic705连接。本发明发光器件用于背光源703,通过终端706提供电流。
由于用本发明发光器件作为上述液晶显示装置的背光源,因此可得到低功耗的背光源。另外,因为本发明发光器件为平面发光器件且可扩大其面积,背光源也可具有大面积。因此,可得到低功耗的较大面积液晶显示装置。
图8作为照明装置的台灯,显示应用本发明的使用发光器件的实例。图8中所示台灯具有外壳801和光源802,本发明发光器件用作光源802。本发明发光器件具有高发光效率的发光元件,因此可用作低功耗的台灯。
图9作为室内照明装置901,显示应用本发明的使用发光器件的实例。因为本发明发光器件的面积也可扩大,本发明发光器件可用作具有大面积的照明装置。另外,本发明发光器件具有高发光效率的发光元件,因此可用作低功耗的照明装置。当图6a中所述本发明电视机902置于房间,其中施用本发明的发光器件用作室内照明装置901时,可观看公共广播和电影。
注意,适当时,可使实施方案方式5与实施方案方式1-4中所述任何结构组合。
[实施方案1]
在实施方案1中,将具体描述结构式(5)代表的本发明咔唑衍生物4-苯基-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp)的合成方法。
[步骤1:4-溴-二苯胺的合成]
在以下(d-1)中显示步骤1中4-溴-二苯基胺的合成流程。
在1-l锥形瓶中,将51g(0.3mol)二苯胺溶于700ml乙酸乙酯后,将54g(0.3mol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。约300小时后,将该混合物溶液用水洗涤,然后向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将滤液浓缩,收集。因此,得到70g深棕色油状目标产物,收率94%。
[步骤2-1:3-溴-9-苯基-9h-咔唑的合成]
以下(d-2-1)显示步骤2-1中3-溴-9-苯基-9h-咔唑的合成流程。
在1000ml锥形瓶中,加入24g(100mmol)9-苯基-9h-咔唑、18g(100mmol)n-溴琥珀酰亚胺、450ml甲苯和200ml乙酸乙酯,将混合物在室温下搅拌45小时。将该悬浮液用水洗涤,然后向其中加入硫酸镁,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,干燥。从而得到32g焦糖状目标产物3-溴-9-苯基-9h-咔唑,收率99%。
[步骤2-2:9-苯基-9h-咔唑-3-硼酸的合成]
以下(d-2-2)显示步骤2-2中9-苯基-9h-咔唑-3-硼酸的合成流程。
在500-ml锥形瓶中,将29g(90mmol)3-溴-9-苯基-9h-咔唑和200ml四氢呋喃(tηf)在-78℃下搅拌为溶液。然后,将110ml(69mmol)正-丁基锂(1.57mol/l己烷溶液)滴加到该溶液中,在相同温度下搅拌2小时。另外,将13ml(140mmol)硼酸三甲酯加入该溶液,将溶液在室温下搅拌24小时。
反应完成后,将200ml盐酸(1.0mol/l)加入反应混合物,然后将混合物在室温下搅拌1小时。将该混合物依次用氢氧化钠水溶液和水洗涤,加入硫酸镁,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,向其中加入氯仿和己烷。将混合物用超声波处理。然后,进行重结晶。因此,得到21g目标白色粉末9-苯基-9h-咔唑-3-硼酸,收率80%。
[步骤3:4-(9-苯基-9h-咔唑-3-基)二苯胺(缩写:pcba)的合成]
以下(d-3)显示步骤3中4-(9-苯基-9h-咔唑-3-基)二苯胺(缩写:pcba)的合成流程。
在500-ml三颈瓶中,加入6.5g(26mmol)4-溴二苯胺、7.5g(26mmol)9-苯基-9h-咔唑-3-硼酸和400mg(1.3mmol)三(邻-甲苯基)膦,通过氮气置换烧瓶中气氛。然后,将100ml甲苯、50ml乙醇和14ml碳酸钾溶液(0.2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入67mg(30mmol)乙酸钯(ii)。
将该混合物在100℃回流10小时。回流后,将该混合物的水层用甲苯萃取。然后,将萃取液与有机层合并,然后用饱和盐水溶液洗涤。有机层的水分经硫酸镁除去后,让混合物自然过滤,将得到的滤液浓缩,得到油性浅棕色物质。该油性物质经硅胶柱层析(展开剂,己烷:甲苯=4:6)纯化。纯化后,将得到的白色固体用二氯甲烷/己烷重结晶,得到4.9g目标白色固体,收率45%。
[步骤4:4-苯基-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp)的合成]
以下(d-4)显示步骤4中4-苯基-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp)的合成流程。
在100-ml三颈瓶中,加入2.0g(4.9mmol)4-(9-苯基-9h-咔唑-3-基)二苯胺、1.1g(4.9mmol)4-溴联苯和2.0g(20mmol)叔丁醇钠,通过氮气置换烧瓶中气氛。然后,将50ml甲苯和0.30ml三(叔丁基)膦(10%(重量)己烷溶液)加入该混合物。
在低压下,搅拌下,将混合物脱气。脱气后,向其中加入0.10g二(二亚苄基丙酮)合钯(0)。随后,将该混合物在80℃下搅拌5小时,以使反应。反应后,将甲苯加入反应混合物,通过抽滤,使该悬浮液依次通过硅藻土、氧化铝和florisil过滤,得到滤液。将得到的滤液依次用饱和碳酸钠溶液和饱和盐水溶液洗涤。将硫酸镁加入有机层,将有机层干燥。干燥后,将该混合物抽滤,将硫酸镁除去;从而得到滤液。
将得到的滤液浓缩,经硅胶柱层析纯化。通过依次用作为展开剂的甲苯∶己烷=1∶9混合溶剂、作为另一种展开剂的甲苯∶己烷=3∶7的混合溶剂洗脱,进行硅胶柱层析。将得到的流分浓缩,将得到的固体用氯仿和己烷混合溶剂重结晶,得到2.3g白色粉末状固体,收率84%。
通过列升华(trainsublimation)法,使得到的1.2g白色固体升华纯化。在7.0pa减压下,在3ml/min氩气流速下,在280℃下,进行升华纯化20小时。从而得到1.1g白色固体,收率89%。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,在图10a和10b中显示1hnmr图谱。由测量结果发现,得到由以上结构式(5)代表的本发明咔唑衍生物4-苯基-4′-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp)。
1hnmr(dmso-d,300mhz):δ(ppm)=7.05-7.20(m,7h),7.28-7.78(m,21h),8.34(d,j=7.8hz,1h),8.57(s,1h).
另外,图11a显示pcba1bp(缩写)的甲苯溶液的吸收光谱。另外,图11b显示pcba1bp(缩写)的薄膜吸收光谱。用紫外-可见分光光度计(v-550,由jascocorporation制造)测量。在石英池中测量溶液的光谱。通过在石英基体上将pcba1bp(缩写)蒸气蒸发制备薄膜样品。图11a显示溶液吸收光谱,该光谱通过从被测样品的吸收减去石英池吸收得到;图11b显示薄膜吸收光谱,该光谱通过从被测样品吸收减去石英基体吸收得到。
在图11a和11b中,横轴代表波长(nm),纵轴代表吸收强度(任意单位)。在甲苯溶液的情况下,在约335nm处观察吸收峰;在薄膜的情况下,在约341nm处观察吸收峰。另外,图11a还显示pcba1bp(缩写)的甲苯溶液的发射光谱(激发波长:346nm)。另外,图11b还显示pcba1bp(缩写)的薄膜发射光谱(激发波长:386nm)。在图11a和11b中,横轴代表波长(nm),纵轴代表光发射强度(任意单位)。在甲苯溶液的情况下,最大发射波长为391nm(激发波长:346nm);在薄膜的情况下,最大发射波长为416nm(激发波长:386nm)。
通过循环伏安(cv)测量法检测pcba1bp(缩写)的氧化-还原反应特性。用电化学分析仪(als型600a或600c,由basinc.制造)测量。
至于用于cv测量的溶液,用脱水二甲基甲酰胺(dmf)(由aldrich生产,99.8%,目录号:22705-6)作溶剂,将为支持电解质的高氯酸四-正丁基铵(n-bu4nclo4,tokyochemicalindustryco.,ltd.的产品,目录号t0836)溶于溶剂,使浓度成为100mmol/l。另外,将待测量的目的物也溶于溶剂,使浓度成为2mmol/l。用铂电极(pte铂电极,由basinc.制造)作工作电极,用另一个铂电极(对于vc-3(5cm)为pt对电极,由basinc.生产)作辅助电极,用ag/ag+电极(re7参比电极用于非水溶剂,由basinc.制造)作参比电极。注意,在室温下(20℃-25℃)测量。另外,进行cv测量的扫描速度为0.1v/秒。
(计算相对于真空水平的参比电极的势能)
先计算用于实施方案1的相对于真空水平的参比电极(ag/ag+电极)的势能(ev)。即计算ag/ag+电极的fermi水平。已知二茂铁的甲醇溶液的氧化-还原电位相对于标准氢电极为+0.610v[相对于she](参见:christianr.goldsmith等,j.am.chem.soc,vol.124,no.1,第83-96页,2002)。在另一方面,发现通过用用于实施方案1的参比电极测量的二茂铁的甲醇溶液的氧化-还原电位为+0.11v[相对于ag/ag+]。因此,发现用于实施方案1的参比电极的势能小于标准氢电极0.50[ev]。
此处,还已知相对于真空水平的标准氢电极的势能为(-4.44ev(参见:toshihiroohnishiandtamamikoyama,macromolecularelmaterial,kyoritsushuppan,第64-67页)。如上所述,经计算,用于实施方案1的相对于真空水平的参比电极的势能为-4.44-0.50=-4.94[ev]。
图41显示对于氧化反应特性的cv测量结果。注意,通过依次在(1)0.07v-1.00v范围,(2)1.00v-0.07v范围内,进行相对于参比电极的工作电极电位的扫描步骤,测量氧化反应特性。
先详细描述通过cv测量,计算pcba1bp(缩写)的homo水平。如图41中所示,氧化峰电位epa为0.536v。另外,还原峰电位epc为0.446v。因此,可计算半波电位(epc与epa之间的中间电位)为0.49v。由此显示,可通过0.49v电能[相对于ag/ag+]将pcba1bp(缩写)氧化,该电能对应于homo水平。
此处,如上所述,用于实施方案1的相对于真空水平的参比电极的势能为-4.94[ev]。因此,发现pcba1bp(缩写)的homo水平为-4.94-0.49=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间氧化还原重复(repetition)具有有利特性。
[实施方案2]
在实施方案2中,将具体描述由结构式(6)代表的本发明咔唑衍生物4,4'-二苯基-4"-(9-苯基-9-h-咔唑-3-基)三苯胺(缩写:pcbbi1bp)的合成方法。
[步骤1-1:4-苯基-二苯胺的合成]
以下(e-1-1)显示步骤1-1中4-苯基-二苯胺的合成流程。
在三颈瓶中,将5.2g(2.5mmol)三-叔丁基膦(10%(重量)己烷溶液)加入含20.0g(85.8mmol)4-溴联苯、16.0g(172mmol)苯胺、0.19g(0.86mmol)乙酸钯(ii)和23.7g(172mmol)碳酸钾的脱水二甲苯悬浮液(150ml)中,在120℃下,将其混合物在氮气氛下回流10小时。反应完成后,将反应混合物用水洗涤,分离为有机层和水层,将水层用甲苯萃取。
将以上得到的甲苯层与以上有机层合并,然后用饱和盐水溶液洗涤。然后,向其中加入硫酸镁,将有机层中水分除去。将混合物抽滤,将得到的滤液浓缩。得到的残渣经硅胶柱层析(展开剂为溶剂:甲苯)纯化。将得到的溶液浓缩,从而得到13.5g4-苯基-二苯胺白色固体,收率64%。
[步骤1-2:4,4'-二苯基三苯胺的合成]
以下(e-1-2)显示步骤1-2中4,4'-二苯基三苯胺的合成流程
在100-ml三颈瓶中,加入3.7g(15mmol)4-苯基-二苯胺、3.5g(15mmol)4-溴联苯、2.5g(25mmol)叔丁醇钠和10mg(0.02mmol)二(二亚苄基丙酮)合钯(0),通过氮气置换烧瓶中气氛。然后,将40ml脱水二甲苯加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,向其中加入0.2ml(60mmol)三(叔丁基)膦(10%(重量)己烷溶液)。
然后,将该混合物在120℃下搅拌5小时,使反应。反应后,将甲苯加入反应混合物,依次通过硅藻土、氧化铝和florisil过滤,将该悬浮液抽滤,得到滤液。将得到的滤液依次用饱和碳酸钠溶液和饱和盐水溶液洗涤。将硫酸镁加入得到的有机层,以除去水分。依次通过硅藻土、氧化铝和florisil过滤,将该混合物抽滤,得到滤液。将丙酮和甲醇加入得到的残渣,将残余物用超声波处理,然后重结晶,得到5.4g白色粉末状固体,收率92%。
[步骤1':4,4'-二苯基三苯胺的合成]
除上述步骤1-1和步骤1-2外,也可用步骤1'中所示合成方法合成4,4'-二苯基三苯胺。注意,以下(e-1')显示步骤1'中4,4'-二苯基三苯胺的合成流程。
在200-ml三颈瓶中,加入1.9g(20mmol)苯胺、9.3g(40mmol)4-溴联苯、4.5g(45mmol)叔丁醇钠、0.4g(2.0mmol)乙酸钯(ii)和1.1g(2.0mmol)1,1-二(二苯基膦基)二茂铁(缩写:dppf),通过氮气置换烧瓶中气氛。然后,将70ml脱水二甲苯加入该混合物。在低压下,搅拌下,将该混合物脱气,将混合物在110℃下搅拌3小时,使反应。反应后,将甲苯加入反应混合物,通过抽滤,使该悬浮液依次通过硅藻土、氧化铝和florisil,得到滤液。将得到的滤液依次用饱和碳酸钠溶液和饱和盐水溶液洗涤。将硫酸镁加入得到的有机层,以除去水分。通过抽滤,依次使该混合物通过硅藻土、氧化铝和florisil,将得到的滤液浓缩。将丙酮和己烷加入得到的残渣,将残余物用超声波处理,然后重结晶,得到5.4g白色粉末状固体,收率67%。
[步骤2:4-溴-4',4"-二苯基三苯胺的合成]
通过用上述步骤1-1和步骤1-2或用步骤1'中所示合成方法合成的4,4'-二苯基三苯胺,合成4-溴-4',4"-二苯基三苯胺。注意,以下(e-2)显示步骤2中所示4-溴-4',4"-二苯基三苯胺的合成流程。
在锥形瓶中,将4.0g(10mmol)4,4'-二苯基三苯胺溶于50ml甲苯和50ml乙酸乙酯混合溶剂后,将n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌120小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,重结晶。从而得到4.5g目标白色粉末,收率95%。
[步骤3:4,4'-二苯基-4"-(9-苯基-9-h-咔唑-3-基)三苯胺(缩写:pcbbi1bp)的合成]
以下(e-3)显示步骤3中4,4'-二苯基-4”-(9-苯基-9-h-咔唑-3-基)三苯胺(缩写:pcbbi1bp)的合成流程。
在100-ml三颈瓶中,加入1.5g(3.1mmol)4-溴-4',4"-二苯基三苯胺、0.9g(3.1mmol)9-苯基-9h-咔唑-3-硼酸、50mg(0.023mmol)乙酸钯(ii)和0.050g(0.17mmol)三(邻-甲苯基)膦,通过氮气置换烧瓶中气氛。注意,因为9-苯基-9h-咔唑-3-硼酸的合成方法与实施方案1中所述相似,其描述被引用至其中,因此,在此省略方法描述。将30ml乙二醇二甲醚(dme)和15ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,将该混合物在90℃下搅拌5小时,使反应。
反应后,将乙酸乙酯加入反应混合物,将该悬浮液用饱和碳酸氢钠溶液和饱和盐水溶液洗涤。将硫酸镁加入有机层,将有机层干燥。干燥后,将该混合物抽滤,将硫酸镁除去;从而得到滤液。将得到的滤液浓缩,将甲苯加入得到的固体中,将混合物溶解。然后,通过抽滤,使该溶液依次通过硅藻土、氧化铝和florisil,得到滤液。将得到的滤液浓缩,经硅胶柱层析纯化。依次用作为展开剂的甲苯:己烷=1:9混合溶剂、作为另一种展开剂的甲苯:己烷=3:7混合溶剂洗脱,进行硅胶柱层析。
将得到的流分浓缩,再将得到的固体用二氯甲烷和己烷混合溶剂重结晶,得到1.3g目标白色固体,收率66%。通过列升华法使1.1g得到的白色固体升华纯化。在4ml/min氩气流速下,在305℃下,在7.0pa减压下,进行升华纯化15小时。从而,得到840mg白色固体,收率76%。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图12a和12b中显示1hnmr图谱。由测量结果发现,得到由以上结构式(6)代表的本发明咔唑衍生物4,4'-二苯基-4"-(9-苯基-9-h-咔唑-3-基)三苯胺(缩写:pcbbi1bp)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.25-7.69(m,32h),8.19(d,j=7.3hz,1h),8.35(s,1h).
另外,图13a中显示pcbbi1bp(缩写)的甲苯溶液的吸收光谱。另外,图13b中显示pcbbi1bp(缩写)的薄膜吸收光谱。用紫外-可见分光光度计(v-550,由jascocorporation制造)测量。在石英池中测量溶液的光谱。通过在石英基体上蒸气蒸发pcbbi1bp(缩写)制备薄膜样品。图13a显示通过从被测样品中减去石英池吸收得到的溶液的吸收光谱,图13b显示通过从被测样品中减去石英池吸收得到的薄膜的吸收光谱。在图13a和13b中,横轴代表波长(nm),纵轴代表吸收强度(任意单位)。在甲苯溶液的情况下,在约347nm处观察吸收峰;在薄膜情况下,在约350nm处观察吸收峰。另外,图13a显示pcbbi1bp(缩写)的甲苯溶液的发射光谱(激发波长:358nm)。另外,图13b显示pcbbi1bp(缩写)的薄膜发射光谱(激发波长:366nm)。在图13a和13b中,横轴代表波长(nm),纵轴代表光发射强度(任意单位)。在甲苯溶液的情况下,最大发射波长为399nm(激发波长:358nm),在薄膜的情况下,最大发射波长为417nm(激发波长:366nm)。
通过循环伏安(cv)测量法检测pcbbi1bp(缩写)的氧化-还原反应特性。因为测量方法与实施方案1的相似,省略方法描述。
图42显示对于氧化反应特性的cv测量结果。如图42所示,可读到的氧化峰电位epa为0.521v,可读到的还原峰电位epc为+0.431v。因此,可计算半波电位(epc与epa之间的中间电位)为+0.48v。按照类似于实施方案1的计算,发现pcbbi1bp(缩写)的homo水平为=-5.42[ev]。另外,甚至在100次循环后,氧化峰具有相似值。因此,发现氧化态和中性态之间的氧化还原重复具有有利特性。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量薄膜的结果表明,pcbbi1bp(缩写)的homo水平为-5.34ev。薄膜的吸收光谱的tauc图显示,吸收限为3.15ev。因此,估计固态中能隙为3.15ev,表示pcbbi1bp(缩写)的lumo水平为-2.19ev。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbbi1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为123℃。按该方式,pcbbi1bp(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbbi1bp(缩写)为难以结晶的物质。
[实施方案3]
在实施方案3中,将具体描述结构式(7)代表的本发明咔唑衍生物9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf)的合成方法。
[步骤1:2-溴-9,9-二甲基芴的合成]
以下(f-1)显示步骤1中2-溴-9,9-二甲基芴的合成流程。
在500-ml锥形瓶中,将12.5g(51mmol)2-溴芴、8.5g(51mmol)碘化钾、14.3g(0.50mol)氢氧化钾和250ml二甲亚砜搅拌30分钟。然后,将10ml甲基碘渐渐加到该混合物中。将该混合物在室温下搅拌48小时。反应后,将400ml氯仿加入反应溶液,将混合物搅拌。将该溶液依次用1n盐酸、饱和碳酸钠溶液和饱和盐水溶液洗涤。将硫酸镁加入得到的有机层,以除去水分。
将该混合物抽滤,浓缩。然后,其残余物经硅胶柱层析纯化。依次用作为展开剂的己烷、作为另一种展开剂的乙酸乙酯:己烷=1:5的混合溶剂洗脱,进行硅胶柱层析。将对应流分浓缩,干燥,得到12g棕色油性物质,收率97%。
[步骤2:9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf)的合成]
以下(f-2)显示步骤2中9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf)的合成流程。
在100-ml三颈瓶中,加入2.0g(4.9mmol)4-(9-苯基-9h-咔唑-3-基)二苯胺(缩写:pcba)、1.3g(4.9mmol)2-溴-9,9-二甲基芴和2.0g(20mmol)叔丁醇钠,通过氮气置换烧瓶中气氛。注意,因为pcba(缩写)的合成方法类似于实施方案2中所述方法,所以引用其中的描述;因此,在此省略描述。然后,将50ml甲苯和0.30ml三(叔丁基)膦(10%(重量)己烷溶液)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,向其中加入0.10g二(二亚苄基丙酮)合钯(0)。然后,将混合物在80℃下搅拌5小时,使反应。反应后,将甲苯加入反应混合物,通过抽滤,使该悬浮液依次通过硅藻土、氧化铝和florisil,得到滤液。
将得到的滤液浓缩,经硅胶柱层析纯化。依次用作为展开剂的甲苯:己烷=1:9混合溶剂、作为另一种展开剂的甲苯:己烷=3:7混合溶剂洗脱,进行硅胶柱层析。将得到的流分浓缩,将得到的固体用氯仿和己烷混合溶剂重结晶,得到1.3g目标化合物,收率44%。
通过列升华法,使得到的1.3g浅黄色固体升华纯化。在7.0pa减压下,在3ml/min氩气流速下,在270℃下,进行升华纯化20小时。从而得到1.0g浅黄色固体,收率77%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。以下描述测量结果,图14a和14b中显示1hnmr图谱。由测量结果发现,得到由以上结构式(7)代表的本发明咔唑衍生物9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf)。
1hnmr(dmso-d,300mhz):δ(ppm)=1.39(s,6h)6.98-7.82(m,26h),8.35(d,j=6.8hz,1h),8.57(s,1h).
另外,图15a显示pcbaf(缩写)的甲苯溶液的吸收光谱。另外,图15b显示pcbaf(缩写)的薄膜吸收光谱。用紫外-可见分光光度计(v-550,由jascocorporation制造)测量。在石英池中测量溶液的光谱。通过在石英基体上将pcbaf(缩写)蒸气蒸发制备薄膜样品。图15a显示溶液的吸收光谱,该光谱通过从被测样品的吸收减去石英池吸收得到;图15b显示薄膜吸收光谱,通过从被测样品的吸收减去石英基体的吸收得到该光谱,在图15a和15b中,横轴代表波长(nm),纵轴代表吸收强度(任意单位)。在甲苯溶液的情况下,在约339nm处观察吸收峰;在薄膜的情况下,在约345nm处观察吸收峰。另外,图15a显示pcbaf(缩写)的甲苯溶液的发射光谱(激发波长:347nm)。另外,图15b显示pcbaf(缩写)的薄膜发射光谱(激发波长:370nm)。在图15a和15b中,横轴代表波长(nm),纵轴代表光发射强度(任意单位)。在甲苯溶液的情况下,最大发射波长为394nm(激发波长:347nm);在薄膜的情况下,最大发射波长为404nm(激发波长:370nm)。
通过循环伏安(cv)测量法检测pcbaf(缩写)的氧化-还原反应特性。因为测量方法与实施方案1相似,省略方法描述。
图43显示对于氧化反应特性的cv测量结果。如图43中所示,可读出的氧化峰电位epa为0.481v,可读出的还原峰电位epc为+0.393v。因此,可计算半波电位(epc与epa之间的中间电位)为+0.44v。根据类似于实施方案1的计算,发现pcbaf(缩写)的homo水平=-5.38[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
[实施方案4]
在实施方案4中,将具体描述由结构式(8)代表的本发明咔唑衍生物n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9'-联芴-2-胺(缩写:pcbasf)的合成方法。
[步骤1-1:9-(联苯-2-基)-2-溴芴-9-醇的合成]
以下(g-1-1)显示步骤1-1中9-(联苯-2-基)-2-溴芴-9-醇的合成流程。
在连接滴液漏斗和dimroth冷凝器的100-ml三颈瓶中,加入1.26g(0.052mol)镁,将烧瓶排空。通过加热和搅拌30分钟将镁活化。冷却至室温后,将烧瓶置于氮气流下。然后,向其中加入5ml乙醚和几滴二溴乙烷,使溶于15ml乙醚的11.65g(0.050mol)2-溴联苯从滴液漏斗缓慢滴加到混合物中。滴加完毕后,将混合物回流3小时,制成格氏试剂。
在连接滴液漏斗和dimroth冷凝器的200-ml三颈瓶中,加入11.7g(0.045mol)2-溴-9-芴酮和40ml乙醚。由滴液漏斗向该反应溶液中缓慢滴加合成的格氏试剂。滴加完成后,将混合物回流2小时,然后在室温下搅拌过夜。反应完成后,将溶液用饱和氯化铵溶液洗涤两次,分离为水层和有机层。将得到的水层用乙酸乙酯萃取两次,将该乙酸乙酯溶液和得到的有机层用饱和盐水溶液洗涤。水分经硫酸镁除去后,抽滤,浓缩,得到18.76g9-(联苯-2-基)-2-溴-9-芴醇固体,收率90%。
[步骤1-2:2-溴-螺-9,9'-联芴的合成]
以下(g-1-2)显示步骤1-2中的2-溴-螺-9,9'-联芴的合成流程
在200-ml三颈瓶中,加入18.76g(0.045mol)合成的9-(联苯-2-基)-2-溴-9-芴醇和100ml冰醋酸,向其中加入几滴浓盐酸,将混合物回流2小时。反应完成后,通过抽滤收集沉淀,将沉淀过滤,用饱和碳酸氢钠溶液和水洗涤。将得到的棕色固体用乙醇重结晶,得到10.24g浅棕色粉末状固体,收率57%。
[步骤2:n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9'-联芴-2-胺(缩写:pcbasf)的合成]
以下(g-2)显示步骤2中n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9'-联芴-2-胺(缩写:pcbasf)的合成流程
在100-ml三颈瓶中,加入2.0g(4.9mmol)4-(9-苯基-9h-咔唑-3-基)二苯胺(缩写:pcba)、1.9g(4.9mmol)2-溴-螺-9,9′-联芴和2.0g(20mmol)叔丁醇钠,通过氮气置换烧瓶中气氛。然后,将50ml甲苯和0.30ml三(叔丁基)膦(10%(重量)己烷溶液)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入0.10g二(二亚苄基丙酮)合钯(0)。
然后,将该混合物在80℃下搅拌5小时,使反应。反应后,将甲苯加入反应混合物,通过抽滤,使该悬浮液依次通过硅藻土、氧化铝和florisil过滤,得到滤液。将得到的滤液依次用饱和碳酸钠溶液和饱和盐水溶液洗涤。将硫酸镁加入有机层除去水分后,将该混合物抽滤,将硫酸镁除去,得到滤液。将得到的滤液浓缩,将得到的固体用氯仿和己烷混合溶剂重结晶,得到3.4g白色粉末状固体,收率94%。通过列升化法使得到的2.3g白色固体升化纯化。在7.0pa减压下,在3ml/min氩气流速下,在310℃下,升化纯化20小时。从而得到1.7g白色固体,收率74%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。以下描述测量结果,图16a和16b显示1hnmr图谱。由测量结果发现,得到由以上结构式(8)代表的本发明咔唑衍生物n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9′-联芴-2-胺(缩写:pcbasf)。
1hnmr(cdcl3,300mhz):δ(ppm)=6.61-6.70(m,2h),6.83(d,j=8.3hz,2h),6.88-7.79(m,30h),8.16(d,j=8.3hz,1h),8.26(s,1h).
另外,图17a显示pcbasf(缩写)的甲苯溶液的吸收光谱。另外,图17b显示pcbasf(缩写)的薄膜的吸收光谱。用紫外-可见光分光光度计(v-550,由jascocorporation制造)测量。在石英池中测量溶液的光谱。通过在石英基体上将pcbasf(缩写)蒸气蒸发制备薄膜样品。图17a显示溶液吸收光谱,通过从被测样品的吸收减去石英池吸收得到该光谱;图17b显示薄膜吸收光谱,通过从被测样品的吸收减去石英基体的吸收得到该光谱。在图17a和17b中,横轴代表波长(nm),纵轴代表吸收强度(任意单位)。在甲苯溶液的情况下,在约338nm处观察吸收峰;在薄膜的情况下,在约345nm处观察吸收峰。另外,图17a还显示pcbasf(缩写)的甲苯溶液的发射光谱(激发波长:352nm)。另外,图17b还显示pcbasf(缩写)的薄膜发射光谱(激发波长:371nm)。在图17a和17b中,横轴代表波长(nm),纵轴代表光发射强度(任意单位)。在甲苯溶液的情况下,最大发射波长为396nm(激发波长:352nm),在薄膜的情况下,最大发射波长为427nm(激发波长:371nm)。
通过循环伏安(cv)测量法检测pcbasf(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1,所以省略描述。
图44显示对于氧化反应特性的cv测量结果。如图44所示,可读到的氧化峰电位epa为0.52v,可读到的还原峰电位epc为+0.428v。因此,可计算半波电位(epc与epa之间的中间电位)为+0.47v。按照类似于实施方案1的计算,发现pcbasf(缩写)的homo水平为=-5.41[ev]。另外,甚至在100次循环后,氧化峰具有相似值。因此,发现氧化态和中性态之间的氧化还原重复具有有利特性。
[实施方案5]
在实施方案5中,将描述用在实施方案1-4中合成的本发明咔唑衍生物形成的发光元件2、发光元件3、发光元件4和发光元件5的制备方法,以及它们的元件特性的测量结果。发光元件2用4-苯基-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp)形成;发光元件3用4,4'-二苯基-4"-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbi1bp)形成,发光元件4用9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf)形成;发光元件5用n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9'-联芴-2-胺(缩写:pcbasf)形成。
注意,图18显示实施方案5中发光元件的各元件结构,其中空穴-传输层1512用以上本发明咔唑衍生物形成。另外,用4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)形成作为对比发光元件的发光元件1,用于空穴-传输层1512。为使发光元件1与各发光元件2-5的对比条件相同,在与发光元件2–5的相同基底上形成发光元件1,将发光元件1与发光元件2–5比较。以下显示用于实施方案5的有机化合物的结构式。
首先,通过溅射法将含氧化硅的氧化铟锡沉积在作为玻璃基底的基底1501上,形成第一电极1502。第一电极1502的厚度设定为110nm,面积设定为2mm×2mm。
然后形成el层1503,其中多层堆叠在第一电极1502上。在实施方案5中,el层1503具有结构,其中将作为空穴-注入层的第一层1511、作为空穴-传输层的第二层1512、作为发光层的第三层1513、作为电子-传输层的第四层1514和作为电子-注入层的第五层1515按顺序堆叠。
将具有第一电极1502的基底固定在基底支持器上,在真空蒸发仪中按使第一电极1502的表面面朝下的方式提供,然后将压力减至约10-4pa。然后使4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)和氧化钼(vi)在第一电极1502上共蒸发,由此形成第一层1511,其为空穴-注入层。控制蒸发速度以使作为空穴-注入层的第一层的厚度可为50nm、npb与氧化钼(vi)的重量比可为4:1(=npb:氧化钼)。注意,共蒸发法为其中用多个蒸发源在一个处理室中同时进行蒸发的蒸发法。
然后,通过蒸发法,用电阻加热(resistiveheating)将空穴-传输材料沉积在第一层1511上,使厚度至10nm,形成作为空穴-传输层的第二层1512。注意,在形成发光元件1的情况下,使用4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb);在形成发光元件2的情况下,使用4-苯基-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcba1bp);在形成发光元件3的情况下,使用4,4'-二苯基-4"-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbi1bp);在形成发光元件4的情况下,使用9,9-二甲基-n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-芴-2-胺(缩写:pcbaf);在形成发光元件5的情况下,使用n-苯基-n-[4-(9-苯基-9h-咔唑-3-基)苯基]-螺-9,9'-联芴-2-胺(缩写:pcbasf)。
然后,通过蒸发法,用电阻加热在第二层1512上形成第三层1513,为发光层。通过使9-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(缩写:czpa)与4-(10-苯基-9-蒽基)-4'-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbapa)共蒸发至30nm厚度,形成第三层1513。此处,控制蒸发速度以使czpa与pcbapa的重量比可为1:0.10(=czpa:pcbapa)。
另外,通过蒸发法,用电阻加热使三(8-羟基喹啉根)合铝(iii)(缩写:a1q)沉积在第三层1513上,至厚度为10nm。然后,通过使红菲绕啉(缩写:bphen)沉积在第三层1513至20nm厚度形成第四层1514,为电子-传输层。
然后,通过使氟化锂(lif)沉积在第四层1514上至1nm厚度,形成作为电子-注入层的第五层1515。
最后,通过蒸发法,用电阻加热使铝沉积至200nm厚度,形成第二电极1504,并形成发光元件1-5。
将通过上述方法得到的发光元件1–5放入具有氮气氛的手套式操作箱,以使发光元件密封但不暴露于大气。然后,测量这些发光元件的操作特性。注意,在室温下进行测量(在25℃下保持常压)。
图19显示发光元件1和2的电流密度与亮度特性之间的关系。图20显示发光元件1和2的电压与亮度特性之间的关系。图21显示发光元件1和2的亮度与电流效率特性之间的关系。图22显示发光元件1和2的电压与电流特性之间的关系。
当发光元件2的激励电压为3.4v时,亮度和电流值分别为1277cd/m2和0.79ma。发现,甚至当发光元件2与用于第二层1512的使用npb的发光元件1对比时,用于第二层1512的使用pcba1bp(缩写)的发光元件2也显示较高亮度。另外,还发现,按电流密度或亮度计,电流效率高。
另外,在发光元件2中,由图23中所示发射光谱中观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现在使用本发明pcba1bp(缩写)的发光元件2的结构中实现了有利的载体平衡。
图24显示不间断照明试验的结果,其中通过恒定电流驱动使发光元件2持续燃亮,初始亮度设为1000cd/m2(纵轴表示相对亮度,假设1000cd/m2为100%)。图24中结果显示,甚至160小时后,发光元件2显示仍具有92%的初始亮度,且发现寿命比发光元件1长。因此,可通过施用本发明pcba1bp(缩写)得到寿命长的发光元件。
图25显示发光元件1和3的电流密度与亮度特性之间的关系。图26显示发光元件1和3的电压与亮度特性之间的关系。图27显示发光元件1和3的亮度与电流效率之间的关系。图28显示发光元件1和3的电压与电流特性之间的关系。
当发光元件3的驱动电压为3.4v时,亮度和电流值分别为1328cd/m2和0.78ma。发现,甚至当发光元件3与用npb用于第二层1512的发光元件1对比时,用pcbbi1bp(缩写)用于第二层1512的发光元件3显示较高亮度。另外,发现按电流密度或亮度计,电流效率高。
另外,在发光元件3中,由图29中所示发射光谱观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现在使用本发明pcbbi1bp(缩写)的发光元件3的结构中实现有利的载体平衡。
图30显示发光元件1和4的电流密度与亮度特性之间的关系。图31显示发光元件1和4的电压与亮度特性之间的关系。图32显示发光元件1和4的亮度与电流效率之间的关系。图33显示发光元件1和4的电压与电流特性之间的关系。
当发光元件4的驱动电压为3.8v时,亮度和电流值分别为1328cd/m2和1.08ma。发现,甚至当发光元件4与用npb用于第二层1512的发光元件1对比时,用pcbaf(缩写)用于第二层1512的发光元件4显示较高亮度。
另外,在发光元件4中,从图34中所示发射光谱中观察到,源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现在使用本发明pcbaf(缩写)的发光元件4的结构中实现了有利的载体平衡。
图35显示不间断照明试验的结果,其中通过恒定电流驱动使发光元件4持续燃亮,初始亮度设为1000cd/m2(纵轴表示相对亮度,假设1000cd/m2为100%)。图35中结果显示,甚至在160小时后,发光元件4显示仍具有92%的初始亮度,且发现寿命比发光元件1长。因此,可通过施用本发明pcbaf(缩写)得到寿命长的发光元件。
图36显示发光元件1和5的电流密度与亮度特性之间的关系。图37显示发光元件1和5的电压与亮度特性之间的关系。图38显示发光元件1和5的亮度与电流效率之间的关系。图39显示发光元件1和5的电压与电流特性之间的关系。
当发光元件5的驱动电压为3.8v时,亮度和电流值分别为1398cd/m2和1.11ma。发现,甚至当发光元件5与用npb用于第二层1512的发光元件1对比时,用pcbasf(缩写)用于第二层1512的发光元件5显示较高亮度。另外,发现按电流密度或亮度计,电流效率高。
另外,在发光元件5中,从图40中所示发射光谱中观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现在使用本发明pcbasf(缩写)的发光元件5的结构中实现了有利的载体平衡。
如上所述,发现用本发明咔唑衍生物形成的发光元件2-5显示与发光元件1同等效率水平。因此,发现可通过施用本发明得到具有高效率的发光元件。
另外,作为实施方案5中所示发光元件1的另一种结构,用pcba1bp(缩写)代替在形成第一层1511时使用的npb(缩写),与氧化钼(vi)共蒸发,形成第一层1511。由于这种发光元件1的效率,在约1000cd/m2亮度下的驱动电压和可靠性,得到与发光元件8的那些值相当的有利值。在实施方案10中,通过使用用于空穴-注入层的npb和氧化钼(vi)的共蒸发膜和使用用于空穴-传输层的pcbbinb(缩写)形成发光元件8。当发光元件1的驱动电压为3.8v时,亮度和电流值分别为949cd/m2和0.65ma,经过1500小时驱动后,发光元件1显示64%的初始亮度。
按如此描述,发现pcba1bp(缩写)为有利的材料,其也为空穴-注入材料。另外,还发现也可通过使用用于空穴-注入层的含氧化钼(vi)的共蒸发膜,得到有利的特性。
另外,作为实施方案5中所示发光元件2的另一种结构,用pcba1bp(缩写)代替在形成第一层1511时使用的npb(缩写),与氧化钼(vi)共蒸发,形成第一层1511。利用这种发光元件2的效率,在约1000cd/m2亮度下的驱动电压和可靠性,得到与发光元件8的那些值等同的有利值。在实施方案10中,通过用npb和氧化钼(vi)的共蒸发膜用于空穴-注入层和用pcbbinb(缩写)用于空穴-传输层形成发光元件8。当发光元件2的驱动电压为3.6v时,亮度和电流值分别为843cd/m2和0.53ma,经过1500小时驱动时,发光元件2显示65%的初始亮度。
按如此描述,发现pcba1bp(缩写)为有利的材料,该材料可同时用于作为空穴-注入层的第一层1511和作为空穴-传输层的第二层1512。因此,可容易的制备元件,还可提高材料使用效率。
[实施方案6]
在实施方案6中,将具体描述由结构式(15)代表的本发明咔唑衍生物(联苯-4-基)(苯基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺(缩写:pcta1bp)的合成方法。
[步骤1:4-[n-(联苯-4-基)-n-苯基]氨基苯基硼酸的合成]
以下(η-1)显示步骤1中4-[n-(联苯-4-基)-n-苯基]氨基苯基硼酸的合成流程。
在300-ml三颈瓶中,加入7.0g(18mmol)4-溴-4'-苯基三苯胺,通过氮气置换烧瓶中气氛。然后,向其中加入80ml四氢呋喃(缩写:thf),将混合物在-78℃下搅拌10分钟。然后,通过注射器将13ml(21mmol)正丁基锂己烷溶液(1.63mol/l)滴加到该溶液,将溶液在-78℃下搅拌1小时。搅拌后,将3.5ml(31mmol)硼酸三甲酯加入反应混合物,将混合物在-78℃下搅拌1小时,然后在室温下搅拌24小时。反应后,将100ml1m稀盐酸加入反应溶液,将混合物在室温下搅拌1小时。搅拌后,将该溶液用乙酸乙酯萃取,将有机层用饱和盐水溶液洗涤。洗涤后,将硫酸镁加入有机层,将有机层干燥。干燥后,通过抽滤将硫酸镁除去,得到滤液。将得到的滤液浓缩,用氯仿和己烷混合溶剂重结晶,得到3.6g目标产物,收率56%。
[步骤2:(联苯-4-基)(苯基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺(缩写:pcta1bp)的合成]
以下(η-2)显示步骤2中(联苯-4-基)(苯基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺的合成流程。
在100-ml三颈瓶中,加入2.2g(5.5mmol)4-[n-(联苯-4-基)-n-苯基]氨基苯基硼酸、2.0g(5.5mmol)3-(4-溴苯基)-9-苯基-9h-咔唑、10mg(0.045mmol)乙酸钯(ii)和0.69g(0.23mmol)三(邻-甲苯基)膦,向其中加入10ml碳酸钾溶液(2.0mol/l)和20ml乙二醇二甲醚(缩写:dme)。在低压下,搅拌下,将该混合物脱气,通过氮气置换烧瓶中气氛。将混合物在90℃下搅拌5小时。搅拌后,将甲苯加入反应混合物,将混合物在90℃下加热。
加热后,将该悬浮液分离为有机层和水层。分离后,将有机层用饱和碳酸氢钠溶液和饱和盐水溶液洗涤。将硫酸镁加入有机层,将有机层干燥。通过抽滤,使该混合物依次通过硅藻土、氧化铝和florisil,得到滤液。将得到的滤液浓缩,得到固体。将得到的滤液溶解,经硅胶柱层析纯化。通过依次用作为展开剂的甲苯:己烷=1:9混合溶剂、作为另一种展开剂的甲苯:己烷=2:3混合溶剂洗脱,进行硅胶柱层析。将得到的流分浓缩,将得到的固体溶于氯仿,经高效液相色谱(hplc)(展开剂,氯仿)纯化。将得到的流分浓缩,将得到的固体用氯仿和己烷混合溶剂重结晶,得到1.7g目标白色固体,收率48%。
通过列升化法使得到的1.0g白色固体升化纯化。在7.0pa减压下,在4ml/min氩气流速下,在300℃下,升化纯化15小时。从而得到0.62g白色固体,收率62%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。以下描述测量结果,图45a和45b显示1hnmr图谱。由测量结果发现,得到由以上结构式(15)代表的本发明咔唑衍生物pcta1bp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.02-7.79(m,32h),8.19(d,j=7.3hz,1h),8.39(s,1h)
另外,测量pcta1bp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约349nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约357nm处观察到位于长波长一侧的吸收峰。
另外,测量pcta1bp的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为405nm(激发波长:320nm);在薄膜的情况下,最大发射波长为420nm(激发波长:284nm)。因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcta1bp(缩写)的homo水平为-5.49ev。薄膜吸收光谱的tauc图显示,吸收限为3.10ev。因此,估计固态中能隙为3.10ev,它表示pcta1bp(缩写)的lumo水平为-2.39ev。
通过循环伏安(cv)测量法检测pcta1bp(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现pcta1bp(缩写)的homo水平=-5.48[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcta1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为118℃。按该方式,pcta1bp(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcta1bp(缩写)为难以结晶的物质。
注意,由于用pcta1bp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,pcta1bp(缩写)在实施方案6中按类似于实施方案5的方法合成用于空穴-传输层;因此得到与发光元件8的那些值等同的有利值,发光元件8在实施方案10中用pcbbinb形成。当该发光元件的驱动电压为3.6v时,亮度和电流值分别为1044cd/m2和0.67ma,经过1100小时驱动后,该发光元件显示52%的初始亮度。
[实施方案7]
在实施方案7中,将具体描述由结构式(190)代表的本发明咔唑衍生物二(联苯-4-基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺(缩写:pctbi1bp)的合成方法。
[步骤1:4-[二(联苯-4-基)氨基]苯基硼酸的合成]
以下(i-1)显示步骤1中4-[二(联苯-4-基)氨基]苯基硼酸的合成流程。
在300-ml三颈瓶中,加入6.0g(13mmol)4-溴-4',4"-二苯基三苯胺,通过氮气置换烧瓶中气氛。然后,向其中加入80ml四氢呋喃(缩写:thf),将混合物在-78℃下搅拌10分钟。然后,通过注射器将10ml正丁基锂己烷溶液(1.63mol/l)滴加到该溶液,将溶液在-78℃下搅拌1小时。搅拌后,将2.8ml(25mmol)硼酸三甲酯加入反应混合物,将混合物在-78℃下搅拌1小时,再在室温下搅拌24小时。搅拌后,将约50ml稀盐酸加入反应混合物,将混合物在室温下搅拌30分钟。搅拌后,将乙酸乙酯加入该混合物,进行萃取。萃取后,将有机层用饱和盐水溶液洗涤。然后,将硫酸镁加入有机层,将有机层干燥。干燥后,将该混合物抽滤,得到滤液。将得到的滤液浓缩,用氯仿和己烷混合溶剂重结晶,得到4.8g目标白色粉末状固体,收率86%。
[步骤2:二(联苯-4-基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺(缩写:pctbi1bp)的合成]
以下(i-2)显示步骤2中二(联苯-4-基)[4'-(9-苯基-9h-咔唑-3-基)联苯-4-基]胺的合成流程。
在100-ml三颈瓶中,加入2.0g(4.5mmol)4-[二(联苯-4-基)氨基]苯基硼酸、1.8g(4.5mmol)3-(4-溴苯基)-9-苯基-9h-咔唑、10mg(0.045mmol)乙酸钯(ii)和0.69g(0.23mmol)三(邻-甲苯基)膦,向其中加入10ml碳酸钾溶液(2.0mol/l)和20ml乙二醇二甲醚(缩写:dme)。在低压下,搅拌下,将混合物脱气,通过氮气置换烧瓶中气氛。将该混合物在90℃下搅拌5小时。搅拌后,将甲苯加入反应混合物,将该混合物在90℃下加热。
加热后,将该悬浮液分离为有机层和水层。分离后,将有机层用饱和碳酸氢钠溶液和饱和盐水溶液洗涤。将硫酸镁加入有机层,将有机层干燥。通过抽滤,使该混合物依次通过硅藻土、氧化铝和florisil,得到滤液。将得到的滤液浓缩,得到固体。将得到的滤液溶于甲苯,经硅胶柱层析纯化。用作为展开剂的甲苯洗脱,进行硅胶柱层析。将得到的流分浓缩,将得到的固体用甲苯和己烷混合溶剂重结晶,得到2.4g目标白色固体,收率74%。
通过列升华法,将得到的白色固体升华纯化。在7.0pa减压下,在3ml/min氩气流速下,在340℃下,进行升华纯化20小时。从而得到0.70g白色固体,其理论产量为1.5g,收率46%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。测量结果描述如下,在图46a和46b中显示1hnmr图谱。由测量结果发现,得到由以上结构式(190)代表的本发明咔唑衍生物pctbi1bp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.18-7.83(m,36h),8.21(d,j=7.3hz,1h),8.40(s,1h).
另外,测量pctbi1bp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约350nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约357nm处观察到位于长波长一侧的吸收峰。
另外,测量pctbi1bp(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为410nm(激发波长:320nm);在薄膜的情况下,最大发射波长为447nm(激发波长:340nm)。因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量薄膜的结果表明,pctbi1bp(缩写)的homo水平为-5.50ev。薄膜吸收光谱的tauc图显示,吸收限为3.14ev。因此,估计固态中能隙为3.14ev,它表示pctbi1bp(缩写)的lumo水平为-2.36ev。
通过循环伏安(cv)测量检测pctbi1bp(缩写)的氧化-还原反应特性。因为测量方法与实施方案1的方法相似,所以省略描述。
按照类似于实施方案1的计算,发现pctbi1bp(缩写)的homo水平为=-5.46[ev]。另外,甚至在100次循环后,氧化峰具有相似值。因此,发现氧化态和中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pctbi1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为133℃。按该方式,pctbi1bp(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现pctbi1bp(缩写)为难以结晶的物质。
注意,由于用pctbi1bp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,该pctbi1bp(缩写)在实施方案7中按类似于实施方案5的方式合成用于空穴-传输层;得到与发光元件8的那些值等同的有利值,发光元件8在实施方案10中用pcbbinp形成。当该发光元件的驱动电压为3.6v时,亮度和电流值分别为873cd/m2和0.56ma,当经过110小时驱动后,该发光元件显示80%的初始亮度。
[实施方案8]
在实施方案8中,将具体描述由结构式(343)代表的本发明咔唑衍生物4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbanb)的合成方法。
[步骤1:3-(4-溴苯基)-9-苯基-9h-咔唑的合成]
以下(j-1)显示步骤1中3-(4-溴苯基)-9-苯基-9h-咔唑的合成流程。
在200-ml三颈烧瓶中,加入3.7g(9.9mmol)3-碘-9-苯基-9h-咔唑、2.0g(9.9mmol)4-溴苯基硼酸和0.61g(2.0mmol)三(邻-甲苯基)膦,将50ml乙二醇二甲醚(缩写:dme)和10ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气,脱气后,通过氮气置换烧瓶中气氛。
然后,将0.11g(0.50mmol)乙酸钯(ii)加入该混合物。将该混合物在80℃下搅拌9.5小时。搅拌后,将该混合物冷却至室温,然后用水洗涤两次。将得到的水层用甲苯萃取两次。然后,将萃取液与有机层合并,然后用饱和盐水溶液洗涤。将有机层用硫酸镁干燥,使该混合物自然过滤,然后将滤液浓缩。
将得到的油性物质溶于约20ml甲苯,通过抽滤,使该溶液依次通过硅藻土、氧化铝和florisil。将得到的滤液浓缩,得到的固体经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化,得到1.9g目标白色粉末状固体,收率49%。
[步骤2:4-(1-萘基)二苯胺的合成]
在以下(j-2)中显示步骤2中4-(1-萘基)二苯胺的合成流程。
在200-ml三颈瓶中,加入12g(50mmol)4-溴二苯胺、8.6g(50mmol)1-萘硼酸、22mg(0.1mmol)乙酸钯(ii)和60mg(0.2mmol)三(邻-甲苯基)膦,将50ml甲苯、20ml乙醇和35ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌2小时,使反应。
反应后,将100ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。将该悬浮液浓缩,经硅胶柱层析(展开剂,甲苯:己烷:乙酸乙酯=1:8:1)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到3.0g目标白色粉末,收率20%。
[步骤3:4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbanb)的合成]
以下(j-3)显示步骤3中4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺的合成流程。
在50-ml三颈瓶中,加入1.2g(3.0mmol)3-(4-溴苯基)-9-苯基-9h-咔唑、0.9g(3.0mmol)4-(1-萘基)二苯胺、0.5g(5.0mmol)叔丁醇钠和6.0mg(0.01mmol)二(二亚苄基丙酮)合钯(0),将15ml脱水二甲苯加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入0.06ml(0.03mmol)三(叔丁基)膦(10%(重量)己烷溶液)。在氮气氛下,将该混合物在120℃下搅拌4.5小时,使反应。
反应后,将250ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶、氧化铝和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.5g目标白色粉末,收率82%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.34;3-(4-溴苯基)-9-苯基-9h-咔唑的为0.46;4-(1-萘基)二苯胺的为0.25。
通过核磁共振法(1hnmr)测量通过以上步骤3得到的化合物。测量结果描述如下,在图47a和47b中显示1hnmr图谱。由测量结果发现,得到由以上结构式(343)代表的本发明咔唑衍生物pcbanb(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.07(t,j=6.6hz,1h),7.25-7.67(m,26h),7.84(d,j=7.8hz,1h),7.89-7.92(m,1h),8.03-8.07(m,1h),8.18(d,j=7.8hz,1h),8.35(d,j=0.9hz,1h).
另外,测量pcbanb(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约335nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约341nm处观察到位于长波长一侧的吸收峰。
另外,测量pcbanb的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为410nm(激发波长:345nm);在薄膜的情况下,最大发射波长为433nm(激发波长:341nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcbanb(缩写)的homo水平为-5.44ev。薄膜吸收光谱的tauc图显示,吸收限为3.25ev。因此,估计固态中能隙为3.25ev,它表示pcbanb(缩写)的lumo水平为-2.19ev。
通过循环伏安(cv)测量检测pcbanb(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现pcbanb(缩写)的homo水平=-5.44[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbanb(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为115℃。按该方式,pcbanb(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbanb(缩写)为难以结晶的物质。
另外,图56-59显示对于发光元件6的元件特性的测量结果,该元件用用于空穴-传输层的本发明咔唑衍生物pcbanb(缩写)形成,在实施方案8中,按类似于实施方案5的方法合成pcbanb。发现,甚至当用于发光元件6的本发明空穴-传输材料与发光元件1的npb对比时,用于发光元件6的本发明空穴-传输材料还显示较高亮度。注意,按类似于实施方案5的方式,用用于空穴-传输层151的4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)形成发光元件1,其作为对比发光元件。
另外,在发光元件6中,由图59中所示发射光谱中观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现本发明空穴-传输材料在发光元件6结构中实现了有利载体平衡。
图60显示不间断照明试验的结果,其中通过恒定电流驱动使发光元件6持续燃亮,初始亮度设为1000cd/m2(纵轴表示相对亮度,假设1000cd/m2为100%)。由图60中结果发现,发光元件6的寿命比发光元件1长。因此,可通过施用本发明得到长寿命的发光元件。
[实施方案9]
在实施方案9中,将具体描述由结构式(229)代表的本发明咔唑衍生物4,4'-二(1-萘基)-4"-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbnbb)的合成方法。
[步骤1:4,4'-二溴三苯胺的合成]
以下(k-1)显示步骤1中4,4'-二溴三苯胺的合成流程。
在500-ml锥形瓶中,将12g(50mmol)三苯胺溶于250ml乙酸乙酯混合溶剂后,将18g(100mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌24小时。反应完成后,将混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,干燥,得到20g目标白色固体,收率99%。
[步骤2:4,4'-二(1-萘基)三苯胺的合成]
以下(k-2)显示步骤2中4,4'-二(1-萘基)三苯胺的合成流程。
在100-ml三颈瓶中,加入6.0g(15mmol)4,4'-二溴三苯胺、5.2g(30mmol)1-萘硼酸、2.0mg(0.01mmol)乙酸钯(ii)和6.0mg(0.02mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和20ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将该混合物在90℃下搅拌4.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到6.4g目标白色粉末,收率86%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.53,4,4'-二溴三苯胺的rf值为0.69。
[步骤3:4-溴-4',4"-二(1-萘基)三苯胺的合成]
以下(k-3)显示步骤3中4-溴-4',4"-二(1-萘基)三苯胺的合成流程。
在300-ml锥形瓶中,将6.4g(13mmol)4,4'-二(1-萘基)三苯胺溶于150ml乙酸乙酯后,将2.3g(13mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌24小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,经硅胶柱层析(展开剂,甲苯:己烷=1:5)纯化。因此,得到1.6g目标白色粉末,收率22%。
[步骤4:4,4'-二(1-萘基)-4"-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbnbb)的合成]
以下(k-4)显示步骤4中4,4'-二(1-萘基)-4"-(9-苯基-9h-咔唑-3-基)-三苯胺的合成流程。
在50-ml三颈瓶中,加入1.4g(2.5mmol)4-溴-4',4"-二(1-萘基)三苯胺、0.7g(2.5mmol)9-苯基-9h-咔唑-3-基-硼酸、4.0mg(0.02mmol)乙酸钯(ii)、6.0mg(0.02mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和2.5ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到0.4g目标白色粉末,收率22%。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图48a和48b显示1hnmr图谱。由测量结果发现,得到由以上结构式(229)代表的本发明咔唑衍生物pcbnbb(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.28-7.72(m,30h),7.85(d,j=7.8hz,2h),7.90-7.93(m,2h),8.06-8.09(m,2h),8.19(d,j=7.5hz,1h),8.38(d,j=1.5hz,1h).
另外,测量pcbnbb(缩写)的吸收光谱(测量范围:200nm-800nm)。在甲苯溶液的情况下,在约345nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约355nm处观察到位于长波长一侧的吸收峰。
另外,测量pcbnbb的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为413nm(激发波长:355nm);在薄膜的情况下,最大发射波长为428nm(激发波长:370nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcbnbb(缩写)的homo水平为-5.46ev。薄膜吸收光谱的tauc图显示,吸收限为3.15ev。因此,估计固态中能隙为3.15ev,它表示pcbnbb(缩写)的lumo水平为-2.31ev。
通过循环伏安(cv)测量检测pcbnbb(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现pcbnbb(缩写)的homo水平=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbnbb(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为136℃。按该方式,pcbnbb(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbnbb(缩写)为难以结晶的物质。
另外,图56-59显示对于发光元件7的元件特性的测量结果,该元件用用于空穴-传输层的本发明咔唑衍生物pcbnbb(缩写)形成,在实施方案9中,按类似于实施方案5的方法合成该咔唑衍生物pcbnbb。发现,甚至当用于发光元件7的本发明空穴-传输材料与发光元件1的npb对比时,用于发光元件7的本发明空穴-传输材料还显示较高亮度。注意,按类似于实施方案5的方式,用用于空穴-传输层151的4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)形成发光元件1,作为对比发光元件。
另外,在发光元件7中,由图59中所示发射光谱中观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现本发明空穴-传输材料在发光元件7结构中实现有利的载体平衡。
图60显示不间断照明试验的结果,其中通过恒定电流驱动使发光元件7持续燃亮,初始亮度设为1000cd/m2(纵轴表示相对亮度,假设1000cd/m2为100%)。由图60中结果发现,发光元件7的寿命比发光元件1长。
[实施方案10]
在实施方案10中,将具体描述由结构式(220)代表的本发明咔唑衍生物4-(1-萘基)-4'-苯基-4"-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbinb)的合成方法。
[步骤1:4-苯基三苯胺的合成]
以下(l-1)显示步骤1中4-苯基三苯胺的合成流程。
在300-ml三颈瓶中,加入9.3g(40mmol)4-溴苯基、6.8g(40mmol)二苯胺、5.0g(50mol)叔丁醇钠和10mg二(二亚苄基丙酮)合钯(0),通过氮气置换烧瓶中气氛。然后,将100ml二甲苯和0.6ml三(叔丁基)膦(10%(重量)己烷溶液)加入该混合物。
在低压下,搅拌下,将该混合物脱气。通过氮气置换气氛后,将混合物在130℃下搅拌3.5小时。搅拌后,将250ml甲苯加入反应混合物,使该悬浮液依次通过硅藻土、氧化铝和florisil过滤。将得到的滤液用水洗涤,干燥,向其中加入硫酸镁。使该混合物依次通过硅藻土、氧化铝和florisil过滤,得到滤液。将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到11g目标白色粉末,收率89%。
[步骤2:4-溴-4'-苯基三苯胺的合成]
以下(l-2)显示步骤2中4-溴-4'-苯基三苯胺的合成流程。
在500-ml锥形瓶中,加入6.4g(20mmol)4-苯基三苯胺、250ml乙酸乙酯和150ml甲苯,将混合物搅拌,然后将3.6g(20mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物搅拌27.5小时。将得到的悬浮液用水洗涤后,水分经硫酸镁除去。将悬浮液浓缩,干燥,得到7.7g目标白色粉末,收率96%。
[步骤3:4-(1-萘基)-4'-苯基三苯胺的合成]
以下(l-3)显示步骤3中4-(1-萘基)-4'-苯基三苯胺的合成流程。
在100-ml三颈瓶中,加入8.0g(20mmol)4-溴-4'-苯基三苯胺、3.4g(20mmol)1-萘硼酸、44mg(0.2mmol)乙酸钯(ii)和60mg(0.4mmol)三(邻-甲苯基)膦,将20ml甲苯、10ml乙醇和15ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将混合物在90℃搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到8.6g目标白色粉末,收率97%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.43,4-溴-4'-苯基三苯胺的rf值为0.50。
[步骤4:4-溴-4'-(1-萘基)-4"-苯基-三苯胺的合成]
以下(l-4)显示步骤4中4-溴-4'-(1-萘基)-4"-苯基-三苯胺的合成流程。
在300-ml锥形瓶中,将8.6g(19mmol)4-(1-萘基)-4'-苯基三苯胺溶于150ml乙酸乙酯后,将3.4g(19mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌24小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到8.1g目标白色粉末,收率80%。
[步骤5:4-(1-萘基)-4'-苯基-4"-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbinb)的合成]
以下(l-5)显示步骤5中4-(1-萘基)-4'-苯基-4"-(9-苯基-9h-咔唑-3-基)三苯胺的合成流程。
在50-ml三颈瓶中,加入1.6g(3.0mmol)4-溴-4′-(1-萘基)-4″-苯基-三苯胺、0.9g(30mmol)9-苯基-9h-咔唑-3-基-硼酸、12mg(0.06mmol)乙酸钯(ii)和36mg(0.12mmol)三(邻-甲苯基)膦,将15ml甲苯、15ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌2小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯∶己烷=1∶4)纯化。将得到的流分浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到0.9g目标白色粉末,收率44%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.26,4-溴-4′-(1-萘基)-4″-苯基-三苯胺的rf值为0.45。
通过核磁共振法(1hnmr)测量通过以上步骤5得到的化合物。测量结果描述如下,图49a和49b显示1hnmr图谱。由测量结果发现,得到由以上结构式(220)代表的本发明咔唑衍生物pcbbinb(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.27-7.69(m,31h),7.84(d,j=7.8hz,1h),7.89-7.92(m,1h),8.04-8.08(m,1h),8.18(d,j=7.8hz,1h),8.36(d,j=1.5hz,1h).
另外,测量pcbbinb(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约342nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约351nm处观察到位于长波长一侧的吸收峰。
另外,测量pcbbinb的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为409nm(激发波长:355nm);在薄膜的情况下,最大发射波长为433nm(激发波长:336nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcbbinb(缩写)的homo水平为-5.35ev。薄膜吸收光谱的tauc图显示,吸收限为3.18ev。因此,估计固态中能隙为3.18ev,它表示pcbbinb(缩写)的lumo水平为-2.17ev。
通过循环伏安(cv)测量检测pcbbinb(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现pcbbinb(缩写)的homo水平=-5.42[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbbinb(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为143℃。按该方式,pcbbinb(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现pcbbinb(缩写)为难以结晶的物质。
另外,图56-59显示对于发光元件8的元件特性的测量结果,该元件用用于空穴-传输层的本发明咔唑衍生物pcbbinb(缩写)形成,在实施方案10中,按类似于实施方案5的方法合成pcbbinb。发现,甚至当用于发光元件8的本发明空穴-传输材料与发光元件1的npb对比时,用于发光元件8的本发明空穴-传输材料也显示较高亮度。注意,按类似于实施方案5的方式,用用于空穴-传输层151的4,4'-二[n-(1-萘基)-n-苯基氨基]联苯(缩写:npb)形成发光元件1,作为对比发光元件。
另外,在发光元件8中,由图59中所示发射光谱中观察到源自pcbapa蓝色发光材料的发射波长,但未观察到源自空穴-传输材料的发射波长。因此,发现本发明空穴-传输材料在发光元件8结构中实现有利载体平衡。
图60显示不间断照明试验的结果,其中通过恒定电流驱动使发光元件8持续燃亮,初始亮度设为1000cd/m2(纵轴表示相对亮度,假设1000cd/m2为100%)。由图60中结果发现,发光元件8的寿命比发光元件1长。因此,可通过施用本发明得到长寿命的发光元件。
另外,作为实施方案10中所示发光元件8的另一种结构,用pcbbinb(缩写)代替在形成第一层1511时使用的npb(缩写),与氧化钼(vi)共蒸发,形成第一层1511。由于这种发光元件8的效率、在约1000cd/m2亮度下的驱动电压和可靠性,得到与发光元件8的那些等同的有利值。在实施方案10中,通过使用用于空穴-注入层的npb和氧化钼(vi)的共蒸发膜和使用用于空穴-传输层的pcbbinb(缩写)形成发光元件8。当发光元件8的驱动电压为4.2v时,亮度和电流值分别为1062cd/m2和0.75ma,经过350小时驱动后,该发光元件8显示81%的初始亮度。
按如此描述,发现pcbbinb(缩写)为有利的材料,可同时用于作为空穴-注入层的第一层1511和作为空穴-传输层的第二层1512。因此,可容易的制备元件,也可提高材料的使用效率。
[实施方案11]
在实施方案11中,将具体描述由结构式(355)代表的本发明咔唑衍生物[4'-(1-萘基)联苯-4-基](苯基)[4-(9-苯基-9h-咔唑-3-基)苯基]胺(缩写:pcbant)的合成方法。
[步骤1:4-(4-溴苯基)-4'-苯基-三苯胺的合成]
以下(m-1)显示步骤1中4-(4-溴苯基)-4'-苯基-三苯胺的合成流程。
在500-ml三颈瓶中,加入22g(70mmol)4,4'-二溴联苯、8.5g(50mmol)二苯胺、1.9g(10mmol)碘化亚铜(i)、2.6g(10mmol)18-冠-6-醚、6.9g(50mmol)碳酸钾和50ml1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu),在氮气氛下,将混合物在180℃搅拌37小时。
反应后,将500ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入己烷和甲醇。将混合物用超声波处理,然后重结晶,得到5.3g目标白色粉末,收率27%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.5;4,4'-二溴联苯的rf值为0.59。
[步骤2:[4'-(1-萘基)联苯-4-基]二苯胺的合成]
以下(m-2)显示步骤2中[4'-(1-萘基)联苯-4-基]二苯胺的合成流程。
在100-ml三颈瓶中,加入4.0g(10mmol)4-(4-溴苯基)-4'-苯基-三苯胺、1.7g(10mmol)1-萘硼酸、11mg(0.05mmol)乙酸钯(ii)和15mg(0.05mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和10ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌7小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过硅胶、氧化铝和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过硅胶、氧化铝和硅藻土,得到滤液。将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到3.6g目标白色粉末,收率80%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.58;4-溴苯基-4'-苯基-三苯胺的rf值为0.65。
[步骤3:(4-溴苯基)[4'-(1-萘基)联苯-4-基]苯胺的合成]
以下(m-3)显示步骤3中(4-溴苯基)[4'-(1-萘基)联苯-4-基]苯胺的合成流程。
在200-ml锥形瓶中,将3.6g(8.0mmol)[4'-(1-萘基)联苯-4-基]二苯胺溶于100ml乙酸乙酯后,将1.4g(8.0mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌72小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到3.9g目标白色粉末,收率93%。
[步骤4:[4'-(1-萘基)联苯-4-基](苯基)[4-(9-苯基-9h-咔唑-3-基)苯基]胺(缩写:pcbant)的合成]
以下(m-4)显示步骤4中[4'-(1-萘基)联苯-4-基](苯基)[4-(9-苯基-9h-咔唑-3-基)苯基]胺的合成流程。
在100-ml三颈瓶中,加入1.6g(3mmol)(4-溴苯基)[4′-(1-萘基)联苯-4-基]苯胺、0.8g(3mmol)9-苯基-9h-咔唑-3-硼酸、6.0mg(0.03mmol)乙酸钯(ii)和18mg(0.03mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在80℃下搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯∶己烷=1∶4)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到1.2g目标白色粉末,收率60%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.28;(4-溴苯基)[4′-(1-萘基)联苯-4-基]苯胺的rf值为0.42。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图50a和50b显示1hnmr图谱。由测量结果发现,得到由以上结构式(355)代表的本发明咔唑衍生物pcbant(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.08(t,j=7.5hz,1h),7.20-7.73(m,30h),7.87(d,j=8.1hz,1h),7.92(d,j=7.2hz,1h),8.00(d,j=8.4hz,1h),8.19(d,j=7.8hz,1h),8.35(d,j=1.8hz,1h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量689.30的主峰(模式为es+),确证得到目标pcbant(缩写)。
另外,测量pcbant(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约342nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约351nm处观察到位于长波长一侧的吸收峰。
另外,测量pcbant的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为414nm(激发波长:355nm);在薄膜的情况下,最大发射波长为342nm(激发波长:365nm)。因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以描述省略。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcbant(缩写)的homo水平为-5.38ev。薄膜吸收光谱的tauc图显示,吸收限为3.11ev。因此,估计固态中能隙为3.11ev,它表示pcbant(缩写)的lumo水平为-2.27ev。
通过循环伏安(cv)测量检测pcbant(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现pcbant(缩写)的homo水平=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbant(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为131℃。按该方式,pcbant(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbant(缩写)为难以结晶的物质。
注意,由于用pcbant(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案11中按类似于实施方案5的方法合成用于空穴-传输层的pcbant;因此得到与在实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为1186cd/m2和0.73ma,经过180小时驱动后,该发光元件显示65%的初始亮度。
[实施方案12]
在实施方案12中,将具体描述由结构式(63)代表的本发明咔唑衍生物4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-苯基-三苯胺(缩写:bcba1bp)的合成方法。
[步骤1:9-(联苯-4-基)-9h-咔唑的合成]
以下(n-1)显示步骤1中9-(联苯-4-基)-9h-咔唑的合成流程。
在200-ml三颈瓶中,加入12g(50mmol)4-溴联苯、8.4g(50mmol)咔唑、230mg(1mmol)乙酸钯(缩写:pd(oac)(ii))、1.8g(3.0mmol)1,1-二(二苯基膦基)二茂铁(缩写:dppf)和13g(180mmol)叔丁醇钠,通过氮气置换烧瓶中气氛。然后,将80ml脱水二甲苯加入该混合物。在低压下,搅拌下,将混合物脱气。在氮气氛下,将混合物在120℃下搅拌7.5小时,使反应。
反应完成后,将约600ml热甲苯加入该悬浮液,使混合物依次通过florisil、氧化铝和硅藻土过滤两次。将得到的滤液浓缩,向其中加入己烷。将混合物重结晶,得到14g目标白色粉末,收率87%。
[步骤2:9-(联苯-4-基)-3-溴-9h-咔唑的合成]
以下(n-2)显示步骤2中9-(联苯-4-基)-3-溴-9h-咔唑的合成流程。
在200-ml锥形瓶中,将3.1g(10mmol)9-(联苯-4-基)-9h-咔唑溶于100ml氯仿后,将1.8g(10mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌24小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,干燥,得到3.7g目标白色粉末,收率95%。
[步骤3:[9-(联苯-4-基)-9h-咔唑-3-基]硼酸]的合成
以下(n-3)显示步骤3中[9-(联苯-4-基)-9h-咔唑-3-基]硼酸的合成流程。
在500-ml三颈瓶中,加入8.0g(20mmol)9-(4-联苯)-3-溴-9h-咔唑,通过氮气置换烧瓶中气氛。然后,向其中加入200ml四氢呋喃(缩写:tηf),达到-78℃。然后,将16ml(24mmol)正丁基锂己烷溶液(1.6mol/l)滴加到该混合物溶液中,将溶液搅拌2小时。然后,将4.0ml(40mmol)硼酸三甲酯加入该反应混合物,将混合物在-78℃下搅拌2小时,然后在室温下搅拌18小时。反应后,将50ml1m稀盐酸加入该反应溶液,将混合物搅拌3小时。将该混合物用甲苯萃取,将得到的有机层用饱和盐水溶液洗涤。洗涤后,将硫酸镁加入有机层,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,向其中加入己烷。将混合物用超声波处理,然后重结晶,得到6.6g目标白色粉末,收率91%。
[步骤4:4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-苯基-三苯胺(缩写:bcba1bp)的合成]
以下(n-4)显示步骤4中4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-苯基-三苯胺的合成流程。
在50-ml三颈瓶中,加入1.2g(3.0mmol)4-溴-4'-苯基-三苯胺、1.1g(3.0mmol)[9-(联苯-4-基)-9h-咔唑-3-基]硼酸、6.0mg(0.03mmol)乙酸钯(ii)和18mg(0.06mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.5g目标白色粉末,收率79%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.45;4-溴-4′-苯基-三苯胺的rf值为0.68。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图51a和51b显示1hnmr图谱。由测量结果发现,得到由以上结构式(63)代表的本发明咔唑衍生物bcba1bp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.06(t,j=7.2hz,1h),7.20-7.72(m,29h),7.83(d,j=8.4hz,2h),8.19(d,j=7.8hz,1h),8.35(s,1h)
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量638.27的主峰(模式为es+),确证得到目标bcba1bp(缩写)。
另外,测量pcba1bp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约336nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约342nm处观察到位于长波长一侧的吸收峰。
另外,测量pcba1bp的发射光谱(缩写)(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为394nm(激发波长:350nm);在薄膜的情况下,最大发射波长为408nm(激发波长:301nm)。因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以描述省略。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcba1bp(缩写)的homo水平为-5.48ev。薄膜吸收光谱的tauc图显示,吸收限为3.19ev。因此,估计固态中能隙为3.19ev,它表示pcba1bp(缩写)的lumo水平为-2.29ev。
通过循环伏安(cv)测量检测pcba1bp(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现pcba1bp(缩写)的homo水平=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcba1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为122℃。按该方式,pcba1bp(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现pcba1bp(缩写)为难以结晶的物质。
注意,由于用pcba1bp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案12中按类似于实施方案5的方法合成用于空穴-传输层的pcba1bp;因此得到与在实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为1031cd/m2和0.72ma,经过180小时驱动后,该发光元件显示89%的初始亮度。
[实施方案13]
在实施方案13中,将具体描述由结构式(364)代表的本发明咔唑衍生物4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-(1-萘基)三苯胺(缩写:bcbanb)的合成方法。
[步骤1:4-溴三苯胺的合成]
以下(o-1)显示步骤1中4-溴三苯胺的合成流程。
向含54.0g(220mmol)三苯胺的1.5l乙酸乙酯溶液中加入35.6g(200mmol)n-溴琥珀酰亚胺(缩写:nbs)。然后,将混合物搅拌24小时。将得到的悬浮液浓缩至1l后,将浓缩的悬浮液用含5%乙酸钠的1l水溶液洗涤。洗涤后,将该溶液进一步浓缩至约50ml。然后,将甲醇加入浓缩溶液,使溶液沉淀,将得到的沉淀过滤,干燥,得到46.5g目标白色粉末,收率73%。
[步骤2:4-(1-萘基)三苯胺的合成]
以下(o-2)中显示步骤2中4-(1-萘基)三苯胺的合成流程。
在20ml三颈瓶中,加入9.7g(30mmol)4-溴三苯胺、5.7g(33mmol)1-萘硼酸、67mg(0.3mmol)乙酸钯(ii)和91g(0.3mmol)三(邻-甲苯基)膦,将20ml甲苯、20ml乙醇和20ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌2小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液依次用碳酸氢钠溶液和水洗涤,向其中加入硫酸镁,将滤液干燥。干燥后,使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,干燥,得到11g目标浅黄色固体,收率99%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.48;4-溴三苯胺的rf值为0.55。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。由测量结果发现,得到由以上结构式(364)代表的本发明化合物。
1hnmr(cdcl3,300mhz):δ(ppm)=7.07(t,j=7.5hz,1h),7.22-7.61(m,21h),7.83(d,j=7.8hz,1h),7.88-7.91(m,1h),8.02-8.05(m,1h).
[步骤3:4-溴-4′-(1-萘基)三苯胺的合成]
以下(o-3)显示步骤3中4-溴-4′-(1-萘基)三苯胺的合成流程。
在500-ml回收瓶(recoveryflask)中,将11g(30mmol)4-(1-萘基)三苯胺溶于300ml乙酸乙酯后,将5.3g(30mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌168小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到7.8g目标白色粉末,收率43%。
[步骤4:4-[9-(联苯-4-基)-9h-咔唑-3-基]-4'-(1-萘基)三苯胺(缩写:bcbanb)的合成]
以下(o-4)显示步骤4中4-[9-(联苯-4-基)-9h-咔唑-3-基]-4'-(1-萘基)三苯胺的合成流程。
在100-ml三颈瓶中,加入1.35g(3.0mmol)4-溴-4′-(1-萘基)三苯胺、1.1g(3.0mmol)[9-(联苯-4-基)-9h-咔唑-3-基]硼酸、6.0mg(0.02mmol)乙酸钯(ii)和9.0mg(0.06mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌3小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯∶己烷=1∶4)纯化。将得到的流分浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.0g目标白色粉末,收率50%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.45;4-溴-4′-(1-萘基)三苯胺的rf值为0.66。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图52a和52b显示1hnmr图谱。由测量结果发现,得到由以上结构式(364)代表的本发明咔唑衍生物bcbanb(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.08(t,j=6.9hz,1h),7.28-7.71(m,28h),7.82-7.86(m,3h),7.89-7.92(m,1h),8.04-8.07(m,1h),8.20(d,j=7.8hz,1h),8.37(d,j=1.2hz,1h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量556.52的主峰(模式为es+),确证得到目标bcbanb(缩写)。
另外,测量bcbanb(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约335nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约344nm处观察到位于长波长一侧的吸收峰。
另外,测量bcbanb(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为410nm(激发波长:345nm);在薄膜的情况下,最大发射波长为422nm(激发波长:328nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以描述省略。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,bcbanb(缩写)的homo水平为-5.42ev。薄膜吸收光谱的tauc图显示,吸收限为3.19ev。因此,估计固态中能隙为3.19ev,它表示bcbanb(缩写)的lumo水平为-2.23ev。
通过循环伏安(cv)测量检测bcbanb(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现bcbanb(缩写)的homo水平=-5.45[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测bcbanb(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为130℃。按该方式,bcbanb(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现bcbanb(缩写)为难以结晶的物质。
注意,用bcbanb(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案13中按类似于实施方案5的方法合成用于空穴-传输层的bcbanb;得到与用实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为848cd/m2和0.52ma。
[实施方案14]
在实施方案14中,将具体描述由结构式(366)代表的本发明咔唑衍生物4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-(1-萘基)4"-苯基-三苯胺(缩写:bcbbinb)的合成方法。
[步骤1:4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-(1-萘基)4"-苯基-三苯胺(缩写:bcbbinb)的合成]
以下(p-1)显示步骤1中4-[9-(联苯-4-基)-9h-咔唑-3-基)-4'-(1-萘基)4"-苯基-三苯胺的合成流程。
在100-ml三颈瓶中,加入1.6g(3.0mmol)4-溴-4′-(1-萘基)-4″-苯基-三苯胺、1.1g(3.0mmol)[9-(联苯-4-基)-9h-咔唑-3-基]硼酸、6.0mg(0.03mmol)乙酸钯(ii)和18mg(0.03mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯∶己烷=1∶4)纯化。将得到的流分浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.4g目标白色粉末,收率60%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.26;4-溴-4′-(1-萘基)-4″-苯基-三苯胺的rf值为0.46。
通过核磁共振法(1hnmr)测量通过以上步骤1得到的化合物。测量结果描述如下,图53a和53b显示1hnmr图谱。由测量结果发现,得到由以上结构式(366)代表的本发明咔唑衍生物bcbbinb(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.30-7.71(m,33h),7.82-7.86(m,3h),7.90-7.93(m,1h),8.05-8.08(m,1h),8.20(d,j=7.8hz,1h),8.38(d,j=1.5hz,1h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量765.32的主峰(模式为es+),确证得到目标bcbbinb(缩写)。
另外,测量bcbbinb(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约342nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约351nm处观察到位于长波长一侧的吸收峰。
另外,测量bcbbinb(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为409nm(激发波长:355nm);在薄膜的情况下,最大发射波长为433nm(激发波长:336nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以描述省略。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,bcbbinb(缩写)的homo水平为-5.35ev。薄膜吸收光谱的tauc图显示,吸收限为3.18ev。因此,估计固态中能隙为3.18ev,它表示bcbbinb(缩写)的lumo水平为-2.17ev。
通过循环伏安(cv)测量检测bcbbinb(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现bcbbinb(缩写)的homo水平=-5.42[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测bcbbinb(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为143℃。按该方式,bcbbinb(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现bcbbinb(缩写)为难以结晶的物质。
注意,用bcbbinb(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案14中按类似于实施方案5的方法合成用于空穴-传输层的bcbbinb;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为996cd/m2和0.59ma,经过180小时驱动后,该发光元件显示84%的初始亮度。
[实施方案15]
在实施方案15中,将具体描述由结构式(386)代表的本发明咔唑衍生物4-{9-[4-(1-萘基)苯基]-9h-咔唑-3-基}-4'-苯基-三苯胺(缩写:nbcba1bp)的合成方法。
[步骤1:9-(4-溴苯基)-9h-咔唑的合成]
以下(q-1)显示步骤1中9-(4-溴苯基)-9h-咔唑的合成流程。
在300-ml三颈瓶中,加入56g(240mmol)1,4-二溴苯、31g(180mmol)9h-咔唑、4.6g(24mmol)碘化亚铜(i)、2.1g(8.0mmol)18-冠-6-醚、66g(480mmol)碳酸钾和8ml1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu),在氮气氛下,将混合物在180℃搅拌6小时。
反应后,将该悬浮液过滤,将滤液依次用稀盐酸、饱和碳酸氢钠溶液和饱和盐水溶液洗涤。然后,水分经硫酸镁除去。将该悬浮液过滤,将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=9:1)纯化。将得到的流分浓缩,向其中加入氯仿和己烷。将混合物用超声波处理,然后重结晶,得到21g目标浅棕色片状结晶,收率35%。
[步骤2:9-[4-(1-萘基)苯基]-9h-咔唑]的合成
以下(q-2)显示步骤2中9-[4-(1-萘基)苯基]-9h-咔唑的合成流程。
在100-ml三颈瓶中,加入4.8g(15mmol)9-(4-溴苯基)-9h-咔唑、2.6g(15mmol)1-萘硼酸、2.0mg(0.01mmol)乙酸钯(ii)和6.0mg(0.02mmol)三(邻-甲苯基)膦,将20ml甲苯、10ml乙醇和10ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌9小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到5.0g目标白色粉末,收率90%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.46;9-(4-溴苯基)-9h-咔唑的rf值为0.54。
[步骤3:3-溴-9-[4-(1-萘基)苯基]-9h-咔唑]的合成
以下(q-3)显示步骤3中3-溴-9-[4-(1-萘基)苯基]-9h-咔唑的合成流程。
在300-ml锥形瓶中,将5.0g(14mmol)9-[4-(1-萘基)苯基]-9h-咔唑溶于50ml甲苯和250ml乙酸乙酯混合溶剂后,将2.5g(14mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌168小时。反应完成后,使该混合物溶液依次通过florisil和硅藻土过滤。然后,将得到的滤液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,向其中加入己烷。然后,将混合物用超声波处理,得到6.1g目标白色粉末,收率99%。
[步骤4:9-[4-(1-萘基)苯基]-9h-咔唑-3-硼酸]的合成
以下(q-4)显示步骤4中9-[4-(1-萘基)苯基]-9h-咔唑-3-硼酸的合成流程。
在500-ml三颈瓶中,加入5.0g(14mmol)3-溴-9-[4-(1-萘基)苯基]-9h-咔唑,通过氮气置换烧瓶中气氛。然后,向其中加入200ml四氢呋喃(缩写:tηf),达到-78℃。将11ml(17mmol)正丁基锂己烷溶液(1.6mol/l)滴加到该混合物溶液,将溶液搅拌4小时。然后,将2.7ml(27mmol)硼酸三甲酯加入该反应混合物,将混合物在-78℃下搅拌2小时,再在室温下搅拌16小时。反应后,将50ml1m稀盐酸加入该反应溶液,将混合物搅拌4小时。将该混合物用甲苯萃取,将得到的有机层用饱和盐水溶液洗涤。洗涤后,将硫酸镁加入有机层,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,向其中加入氯仿和己烷。将混合物用超声波处理,然后重结晶,得到3.5g目标白色粉末,收率63%。
[步骤5:4-{9-[4(1-萘基)苯基]-9h-咔唑-3-基}-4'-苯基-三苯胺(缩写:nbcba1bp)的合成]
以下(q-5)显示步骤5中4-{9-[4(1-萘基)苯基]-9h-咔唑-3-基}-4'-苯基-三苯胺的合成流程。
在50-ml三颈瓶中,加入1.0g(2.5mmol)4-溴-4'-苯基-三苯胺、1.0g(2.5mmol)9-[4-(1-萘基)苯基]-9h-咔唑-3-硼酸、4.0mg(0.02mmol)乙酸钯(ii)和6.0mg(0.02mmol)三(邻-甲苯基)膦,将20ml甲苯、5ml乙醇和2.5ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌13小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.2g目标白色粉末,收率70%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.41;4-溴-4′-苯基-三苯胺的rf值为0.62。
通过核磁共振法(1hnmr)测量通过以上步骤5得到的化合物。测量结果描述如下,图54a和54b显示1hnmr图谱。由测量结果发现,得到由以上结构式(386)代表的本发明咔唑衍生物nbcba1bp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.06(t,j=6.6hz,1h),7.21-7.77(m,30h),7.92-7.98(m,2h),8.04-8.08(m,1h),8.22(d,j=7.8hz,1h),8.37(d,j=1.5hz,1h).
另外,测量nbcba1bp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约333nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约340nm处观察到位于长波长一侧的吸收峰。
另外,测量nbcba1bp(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为393nm(激发波长:350nm);在薄膜的情况下,最大发射波长为488nm(激发波长:302nm)。
因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以描述省略。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,nbcba1bp(缩写)的homo水平为-5.53ev。薄膜吸收光谱的tauc图显示,吸收限为3.22ev。因此,估计固态中能隙为3.22ev,它表示nbcba1bp(缩写)的lumo水平为-2.31ev。
通过循环伏安(cv)测量检测nbcba1bp(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。
按照类似于实施方案1的计算,发现nbcba1bp(缩写)的homo水平=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测nbcba1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为132℃。按该方式,nbcba1bp(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现nbcba1bp(缩写)为难以结晶的物质。
注意,用nbcba1bp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案15中按类似于实施方案5的方法合成用于空穴-传输层的nbcba1bp;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为3.6v时,亮度和电流值分别为773cd/m2和0.47ma。
[实施方案16]
在实施方案16中,将具体描述由结构式(395)代表的本发明咔唑衍生物4-[9-(1-萘基)-9h-咔唑-3-基]-4'-苯基-三苯胺(缩写:ncba1bp)的合成方法。
[步骤1:9-(1-萘基)-9h-咔唑的合成]
以下(r-1)显示步骤1中9-(1-萘基)-9h-咔唑的合成流程。
在500-ml三颈瓶中,加入21g(100mmol)1-溴萘、17g(100mmol)咔唑、0.1g(5.0mmol)碘化亚铜(i)、0.7g(2.5mmol)18-冠-6-醚、33g(240mmol)碳酸钾和80ml1,3-二甲基-3,4,5,6-四氢-2(1h)嘧啶酮(缩写:dmpu),在氮气氛下,将混合物在170℃下搅拌6小时。然后,再将10g(50mmol)1-溴萘、2.0g(10mmol)碘化亚铜(i)和2.6g(10mmol)18-冠-6-醚加入该反应混合物,将混合物再在170℃下搅拌7.5小时。然后,再将10g(50mmol)1-溴萘加入该反应混合物,将混合物再在180℃下搅拌6小时。
反应后,将约200ml甲苯和约100ml盐酸(1mol/l)加入该反应混合物,使混合物通过硅藻土过滤。使得到的滤液通过florisil和硅藻土过滤。将得到的滤液分离为有机层和水层。将该有机层依次用盐酸(1mol/l)和水洗涤,加入硫酸镁,以除去水分。使该悬浮液通过florisil和硅藻土过滤。然后,将得到的滤液浓缩,将己烷加入得到的油性物质中,将混合物用超声波处理,然后重结晶,得到22g目标白色粉末,收率75%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.61;1-溴萘的rf值为0.74;咔唑的rf值为0.24。
[步骤2:3-溴-9-(1-萘基)-9h-咔唑的合成]
以下(r-2)显示步骤2中3-溴-9-(1-萘基)-9h-咔唑的合成流程。
在500-ml锥形瓶中,将5.9g(20mmol)9-(1-萘基)-9h-咔唑溶于50ml甲苯和50ml乙酸乙酯的混合溶剂后,将3.6g(20mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌170小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,干燥,得到7.4g目标白色粉末,收率99%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.43;9-(1-萘基)9h-咔唑的rf值为0.35。
[步骤3:9-(1-萘基)9h-咔唑-3-硼酸的合成]
以下(r-3)显示步骤3中9-(1-萘基)9h-咔唑-3-硼酸的合成流程。
在500-ml三颈瓶中,加入3.7g(10mmol)9-(1-萘基)9h-咔唑,通过氮气置换烧瓶中气氛。然后,向其中加入200ml四氢呋喃(缩写:tηf),将混合物在-78℃下搅拌。然后,将7ml(13mmol)正-丁基锂己烷溶液(1.6mol/l)滴加到该混合物溶液中,将溶液搅拌2小时。然后,将2ml(20mmol)硼酸三甲酯加入该反应混合物,将混合物在-78℃下搅拌3小时,再在室温下搅拌16小时。反应后,将50ml1m稀盐酸加入该反应溶液,将混合物搅拌4小时。将该混合物用乙酸乙酯萃取,将得到的有机层用饱和盐水溶液洗涤。洗涤后,将硫酸镁加入有机层,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,向其中加入氯仿和己烷。将混合物用超声波处理,然后重结晶,得到2.6g目标黄色粉末,收率78%。
[步骤4:4-[9-(1-萘基)-9h-咔唑-3-基]-4'-苯基-三苯胺(缩写:ncba1bp)的合成]
以下(r-4)显示步骤4中4-[9-(1-萘基)-9h-咔唑-3-基]-4'-苯基-三苯胺(缩写:ncba1bp)的合成流程。
在50-ml三颈瓶中,加入1.2g(3.0mmol)4-溴-4'-苯基-三苯胺、1.0g(3.0mmol)9-(1-萘基)9h-咔唑-3-硼酸、6.0mg(0.03mmol)乙酸钯(ii)和0.03mg(18mmol)三(邻-甲苯基)膦,将15ml甲苯、5ml乙醇和3ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将该混合物在90℃下搅拌6.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩。经硅胶柱层析(展开剂,甲苯:己烷=1:3)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到0.5g目标白色粉末,收率25%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.34;4-溴-4'-苯基-三苯胺的rf值为0.54。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。以下描述测量结果,图55a和55b显示1hnmr图谱。由测量结果发现,得到由以上结构式(395)代表的本发明咔唑衍生物ncba1bp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.00-7.07(m,3h),7.19-8.00(m,25h),8.03-8.07(m,2h),8.22-8.25(m,1h),8.40(d,j=1.5,1h).
另外,测量ncba1bp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约333nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约340nm处观察到位于长波长一侧的吸收峰。
另外,测量ncba1bp(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为392nm(激发波长:345nm);在薄膜的情况下,最大发射波长为426nm(激发波长:328nm)。因为吸收光谱和发射光谱的测量方法类似于实施方案1的方法,所以省略描述。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,ncba1bp(缩写)的homo水平为-5.44ev。薄膜吸收光谱的tauc图显示,吸收限为3.19ev。因此,估计固态中能隙为3.19ev,它表示ncba1bp(缩写)的lumo水平为-2.25ev。
通过循环伏安(cv)测量检测ncba1bp(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现ncba1bp(缩写)的homo水平=-5.43[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测ncba1bp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为128℃。按该方式,ncba1bp(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现ncba1bp(缩写)为难以结晶的物质。
注意,用ncba1bp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案16中按类似于实施方案5的方法合成用于空穴-传输层的ncba1bp;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为1198cd/m2和0.82ma。
[实施方案17]
在实施方案17中,将具体描述由结构式(422)代表的本发明咔唑衍生物4,4'-二苯基-4"-(6,9-二苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbi1bpiii)的合成方法。
[步骤1:3-溴-6,9-二苯基-9h-咔唑的合成]
以下(s-1)显示步骤1中3-溴-6,9-二苯基-9h-咔唑的合成流程。
在300-ml锥形瓶中,加入4.8g(15mmol)3,9-二苯基-9h-咔唑,将250ml混合溶剂(乙酸乙酯:甲苯=4:1)加入该溶液。然后,将该混合物搅拌30分钟。然后,将2.7g(15mmol)n-溴琥珀酰亚胺(缩写:nbs)逐渐加到该溶液,将溶液搅拌48小时。
搅拌后,将该混合物依次用饱和碳酸氢钠溶液和饱和盐水溶液洗涤。洗涤后,得到的有机层的水分经硫酸镁除去。然后,将该混合物抽滤,将硫酸镁除去,得到滤液。将得到的滤液浓缩,将少量乙醇加入得到的油性物质。然后,将混合物用超声波处理,使固体沉淀。通过抽滤将沉淀固体收集,得到5.4g白色粉末状固体,收率90%。
[步骤2:4,4'-二苯基-4"-(6,9-二苯基-9h-咔唑-3-基)三苯胺(缩写:pcbbi1bpiii)的合成]
以下(s-2)显示步骤2中4,4'-二苯基-4"-(6,9-二苯基-9h-咔唑-3-基)三苯胺的合成流程。
在100-ml三颈瓶中,加入1.7g(3.8mmol)n,n-二(联苯-4-基)氨基苯基-4-硼酸、1.5g(3.8mmol)3-溴-6,9-二苯基-9h-咔唑、8.4mg(0.038mmol)乙酸钯(ii)和0.080mg(0.26mmol)三(邻-甲苯基)膦。然后,将10ml甲苯、2ml乙醇和10ml2m碳酸钾溶液加入该混合物。在低压下将该混合物脱气后,通过氮气置换烧瓶中气氛。将该混合物在100℃下搅拌3小时。
搅拌后,将甲苯加入该反应混合物,将该混合物在50℃下加热,搅拌。使该悬浮液温度降至室温后,将悬浮液分离为有机层和水层。将得到的有机层依次用饱和碳酸钠溶液和饱和盐水溶液洗涤。洗涤后,将硫酸镁加入得到的有机层,以除去水分。将该混合物抽滤,得到滤液。通过抽滤,使得到的滤液通过硅藻土(wakopurechemicalindustries,ltd.,目录号:531-16855)、florisil(wakopurechemicalindustries,ltd.,目录号:540-00135)和氧化铝,得到滤液。将得到的滤液浓缩,经硅胶柱层析纯化。通过依次用甲苯:己烷=1:4混合溶剂作为展开剂、用甲苯:己烷=1:1混合溶剂作为另一种展开剂进行硅胶柱层析。将得到的流分浓缩,得到固体,用氯仿和己烷混合溶剂重结晶,得到2.3g白色粉末状固体,收率87%。
通过列升化法将得到的2.3g白色固体升化纯化。在7.0pa减压下,在4ml/min氩气流速下,在320℃下,升化纯化18小时。得到1.8g白色固体,收率78%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。以下描述测量结果,图61a和61b显示1hnmr图谱。由测量结果发现,得到由以上结构式(422)代表的本发明咔唑衍生物pcbbi1bpiii(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.22-7.77(m,36h),8.38-8.42(m,2h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量714.30的主峰(模式为es+),确证得到目标pcbbi1bpiii(缩写)。
另外,按下述测量pcbbi1bpiii(缩写)的各种物理性质。
另外,测量pcbbi1bpiii(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约348nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约352nm处观察到位于长波长一侧的吸收峰。另外,测量pcbbi1bpiii(缩写)的发射光谱(测量范围:390nm-550nm)。在甲苯溶液的情况下,最大发射波长为397nm(激发波长:358nm);在薄膜的情况下,最大发射波长为439nm(激发波长:369nm)。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量的薄膜结果表明,pcbbi1bpiii(缩写)的homo水平为-5.46ev。薄膜吸收光谱的tauc图显示,吸收限为3.21ev。因此,估计固态中能隙为3.21ev,它表示pcbbi1bpiii(缩写)的lumo水平为-2.25ev。
通过循环伏安(cv)测量法检测pcbbi1bpiii(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现pcbbi1bpiii(缩写)的homo水平=-41[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbbi1bpiii(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为138℃。按该方式,pcbbi1bpiii(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbbi1bpiii(缩写)为难以结晶的物质。
注意,用pcbbi1bpiii(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案17中按类似于实施方案5的方法合成用于空穴-传输层的pcbbi1bpiii;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.2v时,亮度和电流值分别为1070cd/m2和0.75ma,经过360小时驱动后,该发光元件显示74%的初始亮度。
[实施方案18]
在实施方案18中,将具体描述由结构式(423)代表的本发明咔唑衍生物3,3'-二甲基-4"-苯基-4-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcba1bpiv)的合成方法。
[步骤1:3,3'-二甲基-4"-苯基-三苯胺的合成]
以下(t-1)显示步骤1中3,3'-二甲基-4"-苯基-三苯胺的合成流程。
在100-ml三颈瓶中,加入5.8g(25mmol)4-溴联苯、4.9g(25mmol)间,间'-二甲苯胺、3.0(30mmol)叔丁醇钠和140mg(0.25mmol)二(二亚苄基丙酮)合钯(0),通过氮气置换烧瓶中气氛。然后,将50ml脱水二甲苯加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入1.0ml(0.5mmol)三(叔丁基)膦(10%(重量)己烷溶液)。在氮气氛下,将该混合物在130℃下搅拌1.5小时,使反应。
反应后,将80ml甲苯和420ml己烷加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到8.5g目标白色粉末,收率97%。
通过核磁共振法(1hnmr)测量由以上步骤1得到的化合物。
1hnmr(cdcl3,300mhz):δ(ppm)=2.28(s,6h),6.85(d,j=6.9,2h),6.91-6.95(m,4h),7.09-7.18(m,4h),7.29(t,j=7.5,1h),7.38-7.48(m,4h),7.56-7.59(m,2h).
[步骤2:4-溴-3,3′-二甲基-4″-苯基-三苯胺的合成]
以下(t-2)显示步骤2中4-溴-3,3′-二甲基-4″-苯基-三苯胺的合成流程。
在200-ml锥形瓶中,将2.5g(24mmol)3,3′-二甲基-4″-苯基-三苯胺溶于200ml乙酸乙酯后,将4.3g(24mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌48小时。反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。将该混合物溶液过滤,将得到的滤液浓缩,干燥,得到9.1g目标焦糖状固体,收率88%。
[步骤3:3,3′-二甲基-4″-苯基-4-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcba1bpiv)的合成]
以下(t-3)显示步骤3中3,3′-二甲基-4″-苯基-4-(9-苯基-9h-咔唑-3-基)-三苯胺的合成流程。
在300-ml回收瓶中,加入1.7g(4.0mmol)4-溴-3,3'-二甲基-4”-苯基-三苯胺、1.4g(5.0mmol)9-苯基-9h-咔唑-3-硼酸、5.0mg(0.02mmol)乙酸钯(ii)和6.0mg(0.02mmol)三(邻-甲苯基)膦,将30ml甲苯、5ml乙醇和3.5ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌3小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分,使该悬浮液通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入己烷和丙酮。将混合物用超声波处理,然后重结晶,得到1.0g目标白色粉末,收率42%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯∶己烷=1∶10)展开的目标物的rf值为0.51;4-溴-3,3′-二甲基-4″-苯基-三苯胺的rf值为0.62。
通过核磁共振法(1hnmr)测量通过以上步骤3得到的化合物。测量结果描述如下,图62a至62c显示1hnmr图谱。由测量结果发现,得到由以上结构式(423)代表的本发明咔唑衍生物pcba1bpiv(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=2.26(s,3h),2.30(s,3h),6.86(d,j=7.8,1h),6.99-7.59(m,25h),8.09-8.13(m,2h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量591.28的主峰(模式为es+),确认得到目标pcba1bpiv(缩写)。
另外,按下述测量pcba1bpiv(缩写)的各种物理性质。
另外,测量pcba1bpiv(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约325nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约329nm处观察到位于长波长一侧的吸收峰。另外,测量pcba1bpiv(缩写)的发射光谱(测量范围:370nm-550nm)。在甲苯溶液的情况下,最大发射波长为393nm(激发波长:330nm);在薄膜的情况下,最大发射波长为422nm(激发波长:357nm)。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量薄膜的结果表明,pcba1bpiv(缩写)的homo水平为-5.57ev。薄膜吸收光谱的tauc图显示,吸收限为3.36ev。因此,估计固态中能隙为3.36ev,表示pcba1bpiv(缩写)的lumo水平为-2.21ev。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcba1bpiv(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为105℃。按该方式,pcba1bpiv(缩写)具有高玻璃化转变温度和有利耐热性。另外,不存在结晶峰;因此,发现pcba1bpiv(缩写)为难以结晶的物质。
注意,用pcba1bpiv(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案18中,按类似于实施方案5的方式合成用于空穴-传输层的pcba1bpiv;得到与发光元件8的那些值等同的有利值,在实施方案10中,用pcbbinb形成发光元件8。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为924cd/m2和0.61ma。
[实施方案19]
在实施方案19中,将具体描述由结构式(345)代表的本发明咔唑衍生物4,4'-二(2-萘基)-4"-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbnbbβ)的合成方法。
[步骤1:4,4'-二(2-萘基)-三苯胺的合成]
以下(u-1)显示步骤1中4,4'-二(2-萘基)-三苯胺的合成流程。
在300-ml三颈瓶中,加入6.0g(15mmol)4,4'-二溴三苯胺、6.2g(36mmol)2-萘硼酸、16mg(0.1mmol)乙酸钯(ii)和21mg(0.1mmol)三(邻-甲苯基)膦,将50ml甲苯、20ml乙醇和20ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气气氛下,将该混合物在90℃下搅拌4.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,向其中加入己烷。将混合物用超声波处理,然后重结晶,得到5.6g目标白色粉末,收率75%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.53;4,4'-二溴三苯胺的rf值为0.78。
[步骤2:4-溴-4',4"-二(2-萘基)-三苯胺的合成]
以下(u-2)显示步骤2中4-溴-4',4"-二(2-萘基)-三苯胺的合成流程。
在500-ml锥形瓶中,将4.0g(8.0mmol)4,4′-二(2-萘基)-三苯胺溶于200ml甲苯和250ml乙酸乙酯混合溶剂后,将1.4g(8mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌96小时。反应完成后,就该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯∶己烷=1∶4)纯化。将得到的流分浓缩,向其中加入丙酮和己烷。将混合物用超声波处理,然后重结晶,得到3.4g目标白色粉末,收率61%。
通过核磁共振法(1hnmr)测量通过以上步骤2得到的化合物。测量结果描述如下。
1hnmr(cdcl3,300mhz):δ(ppm)=7.09(d,j=8.4,2h),7.24(d,j=7.8,4h),7.40(d,j=8.4,2h),7.47-7.51(m,4h),7.66(d,j=8.1,4h),7.73-7.76(m,2h),7.85-7.93(m,6h),8.03(s,2h).
[步骤3:4,4′-二(2-萘基)-4″-(9-苯基-9h-咔唑-3-基)三苯胺(缩写:pcbnbbβ)的合成]
以下(u-3)显示步骤3中4,4′-二(2-萘基)-4″-(9-苯基-9h-咔唑-3-基)-三苯胺的合成流程。
在50-ml三颈瓶中,加入1.0g(1.7mmol)4-溴-4',4"-二(2-萘基)-三苯胺、0.6g(2.0mmol)9-苯基-9h-咔唑-3-硼酸、2.2mg(1.0μmol)乙酸钯(ii)和3.0mg(10μmol)三(邻-甲苯基)膦,将20ml甲苯、3ml乙醇和2.0ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气气氛下,将该混合物在90℃下搅拌14小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶、氧化铝和硅藻土过滤。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入甲醇、氯仿、丙酮和己烷。将混合物用超声波处理,然后重结晶,得到1.5g目标浅黄色粉末,收率95%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.31;4-溴-4',4"-二(2-萘基)-三苯胺的rf值为0.56。
通过核磁共振法(1hnmr)测量由以上步骤3得到的化合物。测量结果描述如下,图63a和63b显示1hnmr图谱。由测量结果发现,得到由以上结构式(345)代表的本发明咔唑衍生物pcbnbbβ(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.29-7.90(m,34h),8.03(s,2h),8.16(d,j=7.2,1h),8.34(d,j=1.5,1h).
通过tof-ms检测器(watersmicromasslctpremier,由waters制造)测量以上化合物的分子量。用含乙腈和0.1%甲酸溶液的混合物溶液(乙腈与甲酸溶液的混合比例,80/20vol/vol)作溶剂。由此,检测到分子量739.32的主峰(模式为es+),确证得到目标pcbnbbβ(缩写)。
另外,按下述测量pcbnbbβ(缩写)的各种物理性质。
另外,测量pcbnbbβ(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约357nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约366nm处观察到位于长波长一侧的吸收峰。另外,测量pcbnbbβ(缩写)的发射光谱(测量范围:390nm-550nm)。在甲苯溶液的情况下,最大发射波长为415nm(激发波长:360nm);在薄膜的情况下,最大发射波长为449nm(激发波长:376nm)。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量薄膜的结果表明,pcbnbbβ(缩写)的homo水平为-5.36ev。薄膜吸收光谱的tauc图显示,吸收限为3.06ev。因此,估计固态中能隙为3.06ev,它表示pcbnbbβ(缩写)的lumo水平为-2.30ev。
通过循环伏安(cv)测量检测pcbnbbβ(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现pcbnbbβ(缩写)的homo水平=-5.41[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。由此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbnbbβ(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为129℃。按该方式,pcbnbbβ(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbnbbβ(缩写)为难以结晶的物质。
注意,用pcbnbbβ(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案19中按类似于实施方案5的方法合成用于空穴-传输层的pcbnbbβ;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.4v时,亮度和电流值分别为1104cd/m2和0.74ma,经过650小时驱动后,该发光元件显示75%的初始亮度。
[实施方案20]
在实施方案20中,将具体描述由结构式(424)代表的本发明咔唑衍生物4-苯基-4'-(9-苯基-9h-咔唑-3-基)-4"-(9-苯基芴-9-基)-三苯胺(缩写:pcbbiflp)的合成方法。注意,以上化合物为由通式(1)代表的咔唑衍生物,其中r1为氢,r2为苯基,l为0、m为1,n为0,α2为1,4-亚苯基,α4为1,4-亚苯基,ar1为联苯-4-基,ar2为芴-9-基,芴-9-基的第9位被苯基取代。
[步骤1:4-溴-4'-苯基-二苯胺的合成]
以下(v-1)显示步骤1中4-溴-4'-苯基-二苯胺的合成流程。
在1000-ml锥形瓶中,将37g(150mmol)4-苯基-二苯胺溶于400ml乙酸乙酯后,将27g(150mmol)n-溴琥珀酰亚胺(缩写:nbs)加入该溶液。然后,将该混合物在室温下搅拌24小时。
反应完成后,将该混合物溶液用水洗涤,向其中加入硫酸镁,以除去水分。使该混合物溶液依次通过florisil、硅胶、氧化铝和硅藻土过滤,将得到的滤液浓缩,向其中加入甲苯和己烷。将混合物用超声波处理,然后重结晶,得到4.0g目标白色粉末。另外,使在该重结晶时得到的滤液经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入甲醇。将混合物用超声波处理,然后重结晶,得到4.5g目标白色粉末。从而得到总计8.5g目标白色粉末,收率73%。
[步骤2:4-苯基-4'-(9-苯基-9h-咔唑-3-基)-二苯胺的合成]
以下(v-2)显示步骤2中4-苯基-4'-(9-苯基-9h-咔唑-3-基)-二苯胺的合成流程。
在200-ml三颈瓶中,加入16g(50mmol)4-溴-4'-苯基-二苯胺、16g(55mmol)9-苯基-9h-咔唑-3-硼酸、110mg(0.4mmol)乙酸钯(ii)和150mg(0.4mmol)三(邻-甲苯基)膦,将70ml甲苯、5ml乙醇和23ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将该混合物在90℃下搅拌7.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil、氧化铝、硅胶和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入氯仿和甲醇。将混合物用超声波处理,然后重结晶,得到10g目标浅黄色粉末,收率41%。
[步骤3:9-(4-溴苯基)-9-苯基芴的合成]
以下(v-3)显示步骤3中9-(4-溴苯基)-9-苯基芴的合成流程。
在100-ml三颈瓶中,加入1.2g(50mmol)镁,在低压下,将混合物搅拌30分钟,使镁活化。将烧瓶冷却至室温并使其中变为氮气氛环境后,加入几滴二溴乙烷,以确认泡沫形成和热量产生。将溶于10ml乙醚的12g(50mmol)2-溴联苯缓慢滴加到该混合物后,将混合物搅拌,加热回流2.5小时,制成格氏试剂。
在500-ml三颈瓶中,加入10g(40mmol)4-溴二苯酮和100ml乙醚。将预先合成的格氏试剂缓慢滴加到该混合物后,将混合物搅拌,加热回流9小时。
反应后,将该混合物过滤,得到滤液。将得到的滤液溶于150ml乙酸乙酯,向其中加入1n-盐酸溶液,将混合物搅拌2小时。将该溶液的有机层用水洗涤。然后,加入硫酸镁,以除去水分。将该悬浮液过滤,将得到的滤液浓缩,得到蜜饯状物。
在500-ml回收瓶中,加入该蜜饯状物、50ml冰醋酸和1.0ml盐酸,在氮气氛下,将混合物在130℃下搅拌1.5小时,使反应。反应后,将该反应混合物溶液过滤,得到滤液。将得到的滤液依次用水、氢氧化钠水溶液、水和甲醇洗涤,得到11g目标白色粉末,收率69%。
[步骤4:4-苯基-4'-(9-苯基-9h-咔唑-3-基)-4"-(9-苯基芴-9-基)-三苯胺(缩写:pcbbiflp)的合成]
以下(v-4)显示步骤4中4-苯基-4'-(9-苯基-9h-咔唑-3-基)-4"-(9-苯基芴-9-基)-三苯胺的合成流程。
在100-ml三颈瓶中,加入1.2g(3.0mmol)9-(4-溴苯基)-9-苯基芴、1.5g(3.0mmol)4-苯基-4'-(9-苯基-9h-咔唑-3-基)-二苯胺、0.4mg(4.0mmol)叔丁醇钠和17mg(0.03mmol)二(二亚苄基丙酮)合钯(0),通过氮气置换烧瓶中气氛。然后,将20ml脱水二甲苯加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入0.2ml(0.1mmol)三(叔丁基)膦(10%(重量)己烷溶液)。在氮气氛下,将该混合物在130℃下搅拌5.5小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到1.8g目标白色粉末,收率76%。
通过硅胶薄层层析(tlc)(展开剂,乙酸乙酯:己烷=1:10)展开的目标物的rf值为0.35,9-(4-溴苯基)-9-苯基芴的rf值为0.65,4-苯基-4'-(9-苯基-9h-咔唑-3-基)-二苯胺的rf值为0.19。
通过核磁共振法(1hnmr)测量通过以上步骤4得到的化合物。测量结果描述如下,图64a和64b显示1hnmr图谱。由测量结果发现,得到由以上结构式(424)代表的本发明咔唑衍生物pcbbiflp(缩写)。
1hnmr(cdcl3,300mhz):δ(ppm)=7.02(d,j=8.7,2h),7.12(d,j=8.7,2h),7.17-7.64(m,36h),7.77(d,j=6.9,2h).
另外,按下述测量pcbbiflp(缩写)的各种物理性质。
另外,测量pcbbiflp(缩写)(测量范围:200nm-800nm)的吸收光谱。在甲苯溶液的情况下,在约337nm处观察到位于长波长一侧的吸收峰;在薄膜的情况下,在约339nm处观察到位于长波长一侧的吸收峰。另外,测量pcbbiflp(缩写)的发射光谱(测量范围:390nm-550nm)。在甲苯溶液的情况下,最大发射波长为395nm(激发波长:343nm);在薄膜的情况下,最大发射波长为425nm(激发波长:361nm)。
在常压下,用光电分光光度计(ac-2,由rikenkeikico.,ltd.制造)测量薄膜的结果表明,pcbbiflp(缩写)的homo水平为-5.53ev。薄膜吸收光谱的tauc图显示,吸收限为3.28ev。因此,估计固态中能隙为3.28ev,它表示pcbbiflp(缩写)的lumo水平为-2.25ev。
通过循环伏安(cv)测量检测pcbbiflp(缩写)的氧化-还原反应特性。因为测量方法类似于实施方案1的方法,所以省略描述。按照类似于实施方案1的计算,发现pcbbiflp(缩写)的homo水平=-5.42[ev]。另外,甚至在100次循环后,氧化峰仍具有相似值。因此,发现氧化态与中性态之间的氧化还原重复具有有利特性。
另外,用差示扫描量热法(pyris1dsc,由perkinelmerco.,ltd.制造)检测pcbbiflp(缩写)的玻璃化转变温度。根据测量结果,发现玻璃化转变温度为156℃。按该方式,pcbbiflp(缩写)具有高玻璃化转变温度和有利的耐热性。另外,不存在结晶峰;因此,发现pcbbiflp(缩写)为难以结晶的物质。
注意,用pcbbiflp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案20中按类似于实施方案5的方法合成用于空穴-传输层的pcbbiflp;得到与实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.4v时,亮度和电流值分别为1104cd/m2和0.74ma,经过650小时驱动后,该发光元件显示75%的初始亮度。
注意,用pcbbiflp(缩写)形成的发光元件的效率,在约1000cd/m2亮度下的驱动电压和可靠性,在实施方案20中按类似于实施方案5的方法合成用于空穴-传输层的pcbbiflp;得到与在实施方案10中用pcbbinb形成的发光元件8的那些值等同的有利值。当该发光元件的驱动电压为4.0v时,亮度和电流值分别为1171cd/m2和0.65ma,经过360小时驱动后,该发光元件显示74%的初始亮度。
[实施方案21]
在实施方案21中,将具体描述由结构式(343)代表的本发明咔唑衍生物4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbanb)的合成方法,其不同于实施方案8。
[步骤1:1-(4-溴苯基)-萘的合成]
以下(w-1)显示步骤1中1-(4-溴苯基)-萘的合成流程。
在500-ml三颈瓶中,加入46g(160mmol)4-溴碘苯、24g(140mmol)1-萘硼酸、45mg(0.2mmol)乙酸钯(ii)和60mg(0.2mmol)三(邻-甲苯基)膦,将100ml甲苯、20ml乙醇和11ml碳酸钾溶液(2mol/l)加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,在氮气氛下,将混合物在90℃下搅拌4小时,使反应。
反应后,将500ml甲苯加入该反应混合物,使该悬浮液依次通过florisil和硅藻土过滤。将得到的滤液用水洗涤。然后,加入硫酸镁,以除去水分。使该悬浮液依次通过florisil和硅藻土过滤,得到滤液。将得到的滤液浓缩,经硅胶柱层析(展开剂,己烷)纯化。将得到的流分浓缩,得到25g目标无色透明液体,收率62%。
通过硅胶薄层层析(tlc)(展开剂,己烷)展开的目标物的rf值为0.38;4-溴碘苯的rf值为0.57。
[步骤2:4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺(缩写:pcbanb)的合成]
以下(w-2)显示步骤2中4-(1-萘基)-4'-(9-苯基-9h-咔唑-3-基)-三苯胺的合成流程。
在100-ml三颈瓶中,加入2.8g(10mmol)1-(4-溴苯基)-萘、4.1g(10mmol)4-(9-苯基-9h-咔唑-3-基)-二苯胺、1.2g(12mmol)叔丁醇钠和11mg(0.02mmol)二(二亚苄基丙酮)合钯(0),通过氮气置换烧瓶中气氛。然后,将30ml脱水二甲苯加入该混合物。在低压下,搅拌下,将该混合物脱气。脱气后,向其中加入0.1ml(0.06mmol)三(叔丁基)膦(10%(重量)己烷溶液)。在氮气氛下,将混合物在110℃下搅拌6小时,使反应。
反应后,将150ml甲苯加入该反应混合物,使该悬浮液依次通过florisil、硅胶和硅藻土过滤。将得到的滤液浓缩,经硅胶柱层析(展开剂,甲苯:己烷=1:4)纯化。将得到的流分浓缩,向其中加入丙酮和甲醇。将混合物用超声波处理,然后重结晶,得到5.2g目标白色粉末,收率85%。
注意,除另有说明外,在以上本发明实施方案的各合成方法中所述florisil和硅藻土,分别是指使用florisil(wakopurechemicalindustries,ltd.,目录号:540-00135)和硅藻土(wakopurechemicalindustries,ltd.,目录号:531-16855)。
本申请基于日本专利申请序号2007-312509和日本专利申请序号2008-129917,它们分别于2007年12月3日和2008年5月16日提交到日本专利局,其全文内容通过引用结合到本文中。
参考符号说明
101:基底,102,第一电极,103:el层,104:第二电极,111:第一层(空穴-注入层),112:第二层(空穴-传输层),113:第三层(发光层),114:第四层(电子传输层),115:第五层(电子注入层),301:第一电极,302:第二电极,303:第一el层,304:第二el层,305:电荷发生层,401:激励电路部分(源激励电路),402:像素部分,404:密封基底,405:密封剂,407:空间,408:引线,409:fpc(柔性印刷电路),410:元件基底,411:开关tft,412:电流控制tft,413:第一电极,414:绝缘体,416:el层,417:第二电极,418:发光元件,423:n-通道tft,424:p-通道tft,501:基底,502:第一电极,503:第二电极,504:el层,505:绝缘层,506:间隔层,611:外壳,612:支承基座,613:显示器部分,614:扬声器部分,615:视频输入端,621:主体,622:外壳,623:显示器部分,624:键盘,625:外部连接端口,626:指点器,631:主体,632:外壳,633:显示器部分,634:音频输入部分,635:音频输出部分,636:操作键,637:外部连接端口,638:天线,641:主体,642:显示器部分,643:外壳,644:外部连接端口,645.遥控接受部分,646:图像接受部分,647:电池,648:音频输入部分,649:操作键,650:目镜部分,701:外壳,702:液晶层,703:背光源,704:外壳,705:激励ic,706:终端,801:外壳,802:光源,901:照明装置,902:电视机,1501:基底,1502:第一电极,1503:el层,1504:第二电极,1511:第一层,1512:第二层,1513:第三层,1514:第四层,1515:第五层。
- 还没有人留言评论。精彩留言会获得点赞!