一种治疗消化不良的药物水合物及其制备方法与流程
本发明属于医药技术领域,公开了一种治疗消化不良的药物水合物及其制备方法,具体涉及一种盐酸阿考替胺水合物及其制备方法。
背景技术:
盐酸阿考替胺水合物,由日本泽里新药株式会社和安斯特莱斯制药公司联合开发,于2013年6月6日率先在日本上市,商品名为
功能性消化不良是临床上最常见的一种消化系统功能紊乱性疾病。近年来,功能性消化不良发病率逐年升高。欧美的流行病学调查表明,普通人群中有消化不良症状者占19%~41%。在中国,消化不良症状者约占消化内科患者的40%,占正常人群的19%。严重影响人们的日常生活,逐渐成为现代社会所关注的重要问题。
盐酸阿考替胺水合物是全球批准的第一个功能性消化不良的治疗药物。日本的临床试验结果证明,盐酸阿考替胺水合物具有良好的治疗有效性,且无严重不良反应。
目前,已有多篇文献报导了盐酸阿考替胺水合物的制备及应用。如:
cn1063442c、wo9858918/ep0994108/cn1084739c、wo2012077673/cn103237781a/tw201229024a、wo1996036619、cn101006040b、cn102125551a、cn105924406a、cn103387552a、cn103896873a、cn104672163a、cn103665023a、cn101006040b、cn103665023a、cn103709191a、cn103709120a、cn105198832a、cn104045606a、cn200580028537、cn105439977a、cn105753810a、cn105924406a、cn101006040b、cn102040515a、cn106316979a、cn103665023a、cn1032237781a、cn1184471a、cn1261357a、cn10404560a、cn106316979a、cn102030654a,其经检测均为三水合物。
现有技术公开制备方法的文献很多,但是对盐酸阿考替胺水合物的晶型、制备方法及有关晶型的光谱特性研究的不多,因此本领域需要阿考替胺盐酸盐晶型。
cn105753807a公开了一种盐酸阿考替胺化合物,其含水量为9.5%-10.5%,为阿考替胺三水合物,其分别在55%和45%丁酮中经过两次溶解、降温、保温搅拌30分钟,离心甩滤,滤饼以水淋洗,滤干,干燥得到。得到的盐酸阿考替胺化合物单杂≤0.10%,总杂≤0.50%,dsc图谱中存在2个吸热峰,分别在152.11℃处和170.26℃。
cn104447612a和cn104447611a分别公开了一种阿考替胺盐酸盐水合物晶型及其制备方法,本发明得到的阿考替胺盐酸盐,含有三水化合物,该晶型的熔点为225℃—225.5℃,具有的优点:化学纯度99.9%,最大杂质小于1‰,光学纯度高达99.95%ee;稳定性好,尤其是对湿的稳定性好。所述阿考替胺盐酸盐三水化合物晶型的制备方法,通过将阿考替胺盐酸盐加入4-5倍(重量-体积比)水中,向上述水溶液中加入阿考替胺盐酸盐0.5%—1%的二甲基甲酰胺(dmf),搅拌30分钟,过滤,滤液冷却至10℃-15℃,备用,然后,将阿考替胺盐酸盐18-20倍甲基乙基酮—乙醇=5-6:5-4(或丙酮—乙醇=6-7:4-3)的混合液冷却至10℃-15℃,加入上述备用溶液,保温16-20小时,析出结晶,过滤,经干燥得到结晶形态水合物。
cn103980226a公开了盐酸阿考替胺水合物晶型及其制备方法,所述a型结晶盐酸阿考替胺水合物的差热分析图谱(dsc)在70-95℃、140-160℃,160-170℃处有熔化吸热峰(加热速率为10.00℃/min)。dsc-tga迹线有两个失重平台,在30-80℃处失重7%,90-170℃处失重3%,经检测其为三水合物。a型结晶盐酸阿考替胺水合物的方法,包括以下步骤:(1)将盐酸阿考替胺无水物或盐酸阿考替胺水合物置于适量的溶剂中,加热回流溶解;(2)冷却,析出结晶物;(3)过滤分离从步骤(2)中获得的结晶固体,鼓风干燥,于室温或高湿(湿度大于60%)条件下放置,得a型结晶盐酸阿考替胺水合物。
cn104003958a公开了一种b型结晶盐酸阿考替胺水合物及其制备方法。所述b型结晶盐酸阿考替胺水合物的差热分析图谱(dsc)在173-181℃处有熔化吸热峰,优选在175-179℃处有熔化吸热峰(加热速率为10.00℃/min),其dsc-tga迹线在100-190℃处有3.0%-5.0%的失重,经检测其为一水合物。其制备方法为:(1)将盐酸阿考替胺三水合物置于适量的溶剂中,加热回流溶解;(2)冷却,析出结晶物;(3)过滤分离从步骤(2)中获得的结晶固体,鼓风干燥,于室温或高湿(湿度大于60%)条件下放置,得b型结晶盐酸阿考替胺水合物。本发明所制备的b型结晶盐酸阿考替胺水合物晶型稳定、溶解性好,适合药物开发,所用制备方法安全简便、可操作性强。
cn105237493a公开了一种阿考替胺盐酸盐水合物的i晶型,所述阿考替胺盐酸盐一水合物在70℃左右有吸热峰,失重1.41%,为溶剂和表面水,在120.3-144.6℃有吸热峰,失重3.75%,表明其含有一分子水,在151.6-166.0℃熔化。阿考替胺盐酸盐一水合物在高湿气条件下不稳定,应密封隔绝空气保存在室温条件下。制备方法包括:将阿考替胺与有机溶剂混合,以便获得含有阿考替胺和有机溶剂的第一混合物;将第一混合物中加入成盐溶剂进行成盐反应,以便获得含有阿考替胺盐酸盐的第二混合物;将第二混合物过滤,滤饼用有机溶剂洗涤,干燥,以便获得含有阿考替胺盐酸盐粗品的第三混合物;将第三混合物溶解在含水的有机溶剂中,加热溶解后缓慢降温析晶,以便获得阿考替胺盐酸盐水合物晶体的第四混合物;以及从所述第四混合物中分离所述晶体,并在50~90℃对所分离的晶体进行加热,以便获得所述的阿考替胺盐酸盐一水合物。该方法反应条件温和,工艺简单,能高效得到高纯度盐酸阿考替胺一水合物及其固定晶型。
cn105481791a公开了一种盐酸阿考替胺二水合物的晶型及其制备方法与应用,其制备方法为:将盐酸阿考替胺三水合物与乙醇混合回流,制得盐酸阿考替胺溶液;冷却、析出晶体,获得盐酸阿考替胺二水合物的晶型。其化学性质稳定,在影响因素试验中显示,光照、高湿、高温条件下放置10天,该晶型的总杂不高于0.13%,且无明显引湿性,晶型未发生改变。经过6个月的加速实验,该晶型的总杂不高于0.14%,且无明显引湿性,晶型未发生改变。本发明提供的盐酸阿考替胺二水合物晶型的溶解度略高,制得片剂的溶出度略高,特别是在ph值为1.00-1.50的盐酸溶液中,本发明提供晶型制得的片剂溶出度可达78.03%,优于现有晶型的溶出度(72.46%)。并且制备方法简单,不需采用复杂仪器,制备周期较短,所得产物的产率可达83%以上。在使用差示扫描量热技术进行分析时,表现为在升温速率为每分钟5℃的dsc图谱中存在2个吸热峰,分别在104.3℃±1℃处和198.42℃±1℃;tg线出现2个失重阶段,分别为4.5310%、1.5062%。
现有技术制得的盐酸阿考替胺遇水后容易团聚并且表层形成凝胶样水化层,从而阻碍水份进一步进入颗粒内,造成溶解不完全。这一性质导致制剂中出现“崩而不溶”的情况,使溶出度和生物利用度降低。为解决上述问题,常用的方法为大幅增加辅料用量,以增加原料的分散程度,从而使溶出度和生物利用度达到用药需求。但是,大幅增加辅料会使得制剂的外形显著加大,对于单次服用剂量本身已经较高的药物尤其明显。目前上市的阿考替胺片单次服用剂量为100mg,但是其片重达250mg,即活性成分仅占片重的40%左右。
盐酸阿考替胺属于难溶性药物,常常制备成固体制剂给药,而对于晶型药物的固体制剂来说,制剂的稳定性以及溶出度与原料药的晶型有很大的关系,盐酸阿考替胺在结晶时,如果采用不同的溶剂和工艺条件,则其分子在各晶型晶胞的排列数目和位置及点阵形式不一样,形成不同的晶体结构,盐酸阿考替胺多晶型的变化会改变其性质,质量和药效。因此,盐酸阿考替胺的稳定结晶,对于进一步研究该化合物的物理化学性质,研究其药物组合及临床应用,具有十分重要的意义。
化合物多晶型是一种普遍现象,对于晶型的稳定性、吸湿性、溶解性、流动性、溶出度等优良性能一直是一种长远的追求,也是一个难题。发明经过大量的试验研究发现,现有技术的盐酸阿考替胺水合物存在稳定性、吸湿性、溶解性、流动性、溶出度等各种技术难题,现有技术也一直通过研究晶型解决其溶解性、溶出度、吸湿性等各种问题,但是无法达到理想的状态。
本发明经过大量的试验研究,采用新的结晶方法,惊喜地得到了一种新的盐酸阿考替胺水合物晶型,经检测其含有3.5分子水,本发明提供的盐酸阿考替胺水合物纯度高、对湿热稳定性好,经实验惊喜地发现本发明得到的盐酸阿考替胺水合物制得的片剂溶出度显著提高,有利于提高药物的生物利用度。本发明还公开了盐酸阿考替胺水合物的制备方法,该制备方法简单易操作,反应条件温和,收率及纯度高,适合大规模生产。
技术实现要素:
本发明的发明目的在于提出一种治疗消化不良的药物水合物及其制备方法,具体是指一种盐酸阿考替胺水合物及其制备方法。
为了实现本发明的目的,采用的技术方案为:
一种治疗消化不良的药物水合物及其制备方法,其特征在于,所述水合物为盐酸阿考替胺水合物,其分子式为:c21h30n4o5s·hcl·3.5h2o,结构式如式ii所示,其以2θ±0.2°衍射角表示的x-射线粉末衍射图谱在5.2248°、6.1056°、6.6649°、9.3738°、12.3428°、15.1198°、17.2123°、23.3487°、24.1078°、24.4568°、25.2179°、26.0846°、27.1325°、28.7654°和29.9326°处显示有特征衍射峰。
优选地,本发明提供的盐酸阿考替胺水合物,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示。
本发明提供的盐酸阿考替胺水合物,其制备方法包括如下步骤:
(1)在声场作用下,边搅拌边将盐酸阿考替胺粗品加入到甲酰胺、水的混合溶液中,升温搅拌至溶解;
(2)边搅拌边加入正丁醇;
(3)正丁醇加完后,在声场作用下,降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物。
优选地,步骤1)中所述声场频率为25-30khz、输出功率为40-60w。
优选地,步骤1)中所述甲酰胺、水的混合溶液体积为盐酸阿考替胺粗品重量的5-7倍,甲酰胺、水的体积比为3.5:4.5。
优选地,步骤2)中所述正丁醇体积为盐酸阿考替胺粗品重量的2-4倍。
优选地,步骤1)、步骤2)中所述搅拌速度为150-260转/分钟。
优选地,步骤3)中所述声场频率为15-20khz、输出功率为10-20w。
优选地,步骤3)中所述降温速度为2-4℃/小时。
本发明中,所述的盐酸阿考替胺粗品可以为待进一步纯化的盐酸阿考替胺固体混合物,也可以为市售原料或通过现有技术方法制备得到的其他水合物或粗品,所得的晶型结果均在误差范围内,均为本发明新晶型。
晶体的形成机理很复杂,一个新晶体的获得也具有很大的偶然性,有时不同的溶剂、在不同的结晶条件下会产生相同的晶体结构。某些特定晶型也并非一定会获得更加有利的理化性质。药物的稳定性、吸湿性、溶解性、生物活性、毒性等性质会因晶型的不同而产生巨大的差异。
本发明通过选择不同的溶剂溶解及不同的溶剂析晶进行大量的试验筛选,得到了本发明的制备方法,通过对溶剂种类、搅拌速度、溶剂用量、温度及养晶时间的控制,意外的得到了一种盐酸阿考替胺水合物新晶型。本发明提供的盐酸阿考替胺水合物纯度高、对湿热稳定性好,经实验惊喜地发现本发明得到的盐酸阿考替胺水合物溶出度显著提高,有利于提高药物的生物利用度。本发明还公开了盐酸阿考替胺水合物的制备方法,该制备方法简单易操作,反应条件温和,收率及纯度高,适合大规模生产。
研究表明,在x-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。
本发明所提供的盐酸阿考替胺水合物结晶证实含有3.5分子的结晶水,其性状为白色结晶性粉末,在常温干燥条件下不会发生结晶水的流失。且其粉末x-射线衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。
下面通过对本发明提供的盐酸阿考替胺水合物晶型进行研究来解释和说明本发明技术方案:
1、元素分析c21h30n4o5s·hcl·3.5h2o
仪器:varioelcube元素分析仪;测量元素为c、h、o、s、n;
dioenxdx-500型离子色谱仪;测量元素为cl。
元素分析(%)理论值为:h(6.96),c(45.85),n(10.19),o(24.72),s(5.83),cl(6.44)。
元素分析(%)测定值为:h(6.94),c(45.83),n(10.22),o(24.71),s(5.84),cl(6.43)。
与元素分析的理论值基本相符。
2、晶型检测
取本发明制备得到的盐酸阿考替胺水合物,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图在5.2248°、6.1056°、6.6649°、9.3738°、12.3428°、15.1198°、17.2123°、23.3487°、24.1078°、24.4568°、25.2179°、26.0846°、27.1325°、28.7654°和29.9326°处显示有特征峰。
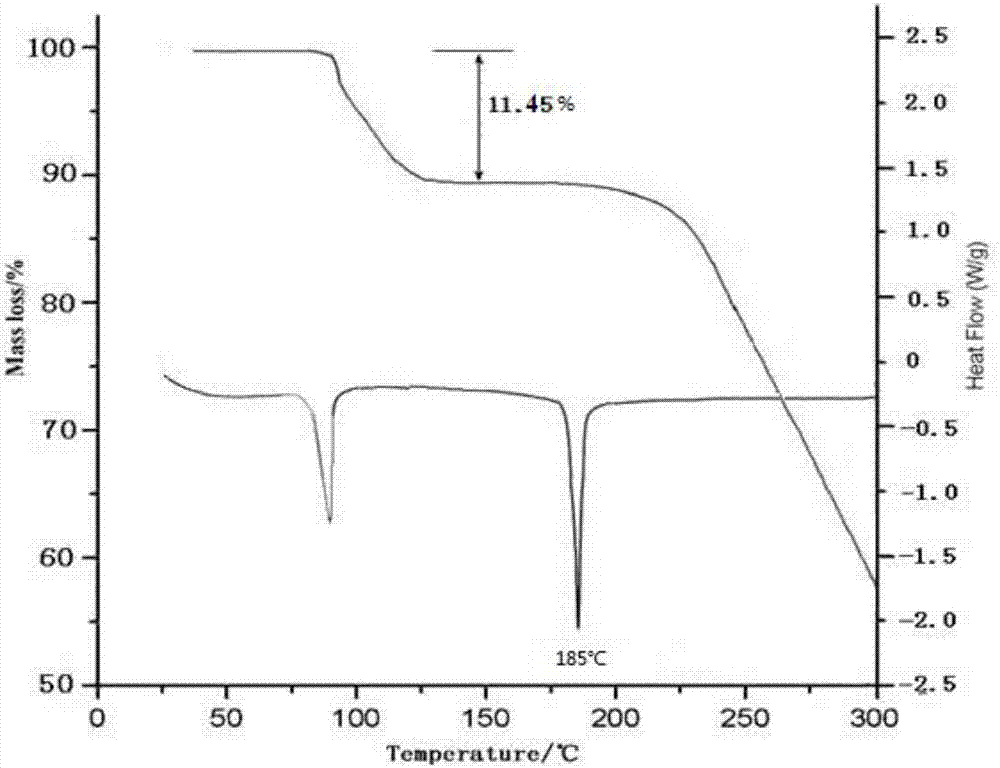
3、差热分析及热重分析
对本发明制备的盐酸阿考替胺晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在100℃左右有吸收峰,快速失去约3.5个水分子的重量,结合元素分析结果,说明该盐酸阿考替胺晶体为3.5水合物;本品在约185℃处有吸热峰;本品经熔点测定:184.0℃-186.0℃。
4、水分分析
采用卡式水分测定仪测定,本发明的盐酸阿考替胺水合物的水含量为11.43-11.50%,与3.5水合物的理论含水量11.46%一致,证明本发明含3.5分子的结晶水。
5、纯度检测
经hplc纯度检测,本发明制备得到的盐酸阿考替胺水合物的纯度可达到99.997~99.999%。
与现有技术相比,本发明具有如下优点:
(1)本发明所提供的盐酸阿考替胺水合物是一种不同于现有技术的新晶型;本发明所提供的盐酸阿考替胺水合物的制备方法简单易操作,反应条件温和,收率大于99.8%,纯度高,适合大规模生产。
(2)本发明提供的盐酸阿考替胺水合物纯度高、对湿热稳定性好,经实验惊喜地发现本发明得到的盐酸阿考替胺水合物制得的片剂溶出度显著提高,有利于提高药物的生物利用度。
附图说明
图1为本发明制备的盐酸阿考替胺水合物的x-射线粉末衍射图谱。
图2为本发明实施例1制备的盐酸阿考替胺水合物的tg-dsc图谱。
具体实施例
以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。
实施例1、盐酸阿考替胺水合物的制备
(1)在频率为30khz、输出功率为50w的声场下,以200转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺粗品重量的6倍的甲酰胺、水的混合溶液中,甲酰胺、水的体积比为3.5:4.5,升温搅拌至溶解;
(2)以200转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺粗品重量3倍的正丁醇;
(3)正丁醇加完后,在频率为20khz、输出功率为15w的声场下,以3℃/小时降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物99.95g,收率99.95%,纯度99.999%。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图见图1,tg-dsc图谱见图2。
实施例2、盐酸阿考替胺水合物的制备
(1)在频率为25khz、输出功率为60w的声场下,以260转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺粗品重量的5倍的甲酰胺、水的混合溶液中,甲酰胺、水的体积比为3.5:4.5,升温搅拌至溶解;
(2)以260转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺粗品重量2倍的正丁醇;
(3)正丁醇溶液加完后,在频率为15khz、输出功率为20w的声场下,以2℃/小时降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物99.90g,收率99.90%,纯度99.998%。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似,tg-dsc图谱与实施例1相似。
实施例3、盐酸阿考替胺水合物的制备
(1)在频率为25khz、输出功率为40w的声场下,以150转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺粗品重量的7倍的甲酰胺、水的混合溶液中,甲酰胺、水的体积比为3.5:4.5,升温搅拌至溶解;
(2)以150转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺粗品重量4倍的正丁醇;
(3)正丁醇溶液加完后,在频率为15khz、输出功率为10w的声场下,以4℃/小时降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物99.88g,收率99.88%,纯度99.999%。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似,tg-dsc图谱与实施例1相似。
实施例4、盐酸阿考替胺水合物的制备
(1)在频率为25khz、输出功率为40w的声场下,以150转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺粗品重量的5倍的甲酰胺、水的混合溶液中,甲酰胺、水的体积比为3.5:4.5,升温搅拌至溶解;
(2)以150转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺粗品重量2倍的正丁醇;
(3)正丁醇溶液加完后,在频率为15khz、输出功率为10w的声场下,以2℃/小时降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物99.87g,收率99.87%,纯度99.998%。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似,tg-dsc图谱与实施例1相似。
实施例5、盐酸阿考替胺水合物的制备
(1)在频率为25khz、输出功率为60w的声场下,以260转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺粗品重量的7倍的甲酰胺、水的混合溶液中,甲酰胺、水的体积比为3.5:4.5,升温搅拌至溶解;
(2)以260转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺粗品重量4倍的正丁醇;
(3)正丁醇溶液加完后,在频率为15khz、输出功率为20w的声场下,以4℃/小时降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物99.92g,收率99.92%,纯度99.999%。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似,tg-dsc图谱与实施例1相似。
下面通过实验例进一步说明本发明:
实验例1:溶剂筛选试验
采用本发明的制备方法操作,具体如下:
(1)在频率为25khz、输出功率为40w的声场下,以150转/分钟的搅拌速度,边搅拌边将盐酸阿考替胺粗品加入到体积为盐酸阿考替胺重量的5倍的溶剂a和溶剂b的混合溶液中,升温搅拌至溶解;
(2)以150转/分钟的搅拌速度,边搅拌边加入体积为盐酸阿考替胺重量2倍的溶剂c;
(3)溶剂c加完后,在频率为15khz、输出功率为10w的声场下,以2℃/小时降温至0-2℃,养晶3-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物。
表1溶剂筛选实验结果
发明人在溶剂筛选过程中,对大多数有机溶剂进行了筛选,不同的组合结晶效果不同,在此仅列举部分筛选试验的数据。
发明人在试验过程中惊喜地发现在甲酰胺与水混合溶剂体系溶解盐酸阿考替胺粗品,并用正丁醇析晶,效果相对于单用其他溶剂体系更好。然后进一步对甲酰胺与水的混合溶剂的比例进行了筛选,发现当甲酰胺与水的体积比大于或小于3.5:4.5时,其收率和纯度稍差一些。当甲酰胺与水体积比为3.5:4.5时,效果是最好的,不仅纯度高、收率高,而且通过实验惊喜地发现其溶出度显著提高,有利于提高药物的生物利用度。故最终确定选择以甲酰胺:水3.5:4.5为溶解溶剂,加正丁醇析晶,以做进一步筛选。
实验例2:结晶试验条件筛选
(1)在声场作用下,边搅拌边将盐酸阿考替胺粗品加入到甲酰胺、水的混合溶液中,升温搅拌至溶解;
(2)边搅拌边加入正丁醇;
(3)正丁醇加完后,在声场作用下,降温至0-2℃,养晶4-6小时,洗涤,真空干燥,得到盐酸阿考替胺水合物。
表2-1结晶试验条件筛选结果
表2-2结晶试验条件筛选结果
表2-3结晶试验条件筛选结果
表2-4结晶试验条件筛选结果
关于结晶试验条件的筛选试验非常复杂,在此我们只列举其中的一部分试验结果。从上述试验结果可以看出,晶体的结晶过程中存在太多的变量,每一个工艺参数的变化都可能对结果产生影响。由实验结果可以看出溶解溶剂用量、析晶溶剂用量、降温速度、结晶时降温速度、声场对产品的收率、纯度及晶体的形成、结晶水的含量等都有较大影响;甲酰胺的加入有助于水合物的形成;而甲酰胺与水的体积比与结晶水的含量有关,体积比大于或小于3.5:4.5均易形成三水合物,溶出度、稳定性都不好。同时,养晶时间、干燥温度、干燥时间等对产品的收率及纯度有较大影响,对晶型的形成影响较小。发明人经过大量试验,最终确定了本发明技术方案的工艺。
实验例3:稳定性试验
1、影响因素实验
将试验品模拟上市包装,在高温60℃、光照5000lx、高湿(rh92.5%)条件下放置10天,按稳定性重点考察项目进行检测,与0天样品比较,结果见表3。有关物质检测参照中国药典2015版有关物质检测方法进行检测。
表3影响因素实验结果
2、加速试验
取实施例1-5制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于6个月末取样测定性状、水分、纯度等,结果见表4。
表4加速试验结果(温度40±2℃,相对湿度75±5%)
从表4看出,本发明盐酸阿考替胺水合物新晶型在温度40±2℃、相对湿度75±5%的条件下放置6个月,各指标均无明显变化,说明本品稳定性好。
3、长期试验
取实施例1-5制备的样品,于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、含量,结果见表5。
表5长期试验结果(温度25±2℃,相对湿度60±5%)
从表5看出,本发明盐酸阿考替胺水合物结晶在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化。说明化学稳定性良好,适合药物制剂的制造及长期储存。
实验例4:溶出度检测试验
溶出度测定方法:将试验品和对照品按照《中华人民共和国药典》2015年版二部附录xc法第二法桨法考察溶出度。以0.01n盐酸900ml为溶出介质,温度为37℃,转速为75r·min-1,每个待测样品中的药物量为30mg。采用紫外分光光度法分别于5、10、15、20、30、45、60min取样进行溶出度测定,检测波长为280nm,在5~25mg·l-1内线性关系良好,回收率、精密度实验均符合方法学要求,结果见表6。
试验品1-6:本发明实施例1-6所制备的样品参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品1:参照专利cn105753807a实施例1制得的盐酸阿考替胺化合物(检测为三水化合物)参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品2:参照专利cn104447612a实施例1制得的盐酸阿考替胺三水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品3:参照专利cn104447611a实施例1制得的盐酸阿考替胺三水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品4:参照专利cn103980226a实施例1-6制得的盐酸阿考替胺三水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品5:参照专利cn104003958a实施例1-2制得的盐酸阿考替胺一水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品6:参照专利cn105237493a实施例1-3制得的盐酸阿考替胺一水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品7:参照专利cn105237493a对比例1-2制得的盐酸阿考替胺三水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
对照品8:cn105481791a实施例1制得的盐酸阿考替胺二水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片;
上述对照品均为经过多次重复实验制得的晶型,并多次进行x射线粉末衍射检测,待晶型稳定(检测结果基本一致)方可作为对照品(如原专利公开了附图,则将测定图与之对比,基本一致方可作为对照品使用)。
对照品9:参照专利cn105412026a实施例2、4、6制得盐酸阿考替胺片;
对照品10:参照专利cn106074407a实施例1-4制得盐酸阿考替胺片;
对照品11:参照专利cn105919967a实施例4、5制得盐酸阿考替胺片;
对照品12:参照专利cn106344517a实施例1制得盐酸阿考替胺片;
对照品13:参照专利cn105769784a实施例1-7制得盐酸阿考替胺片;
对照品14:参照专利cn105769784a实施例8-10制得盐酸阿考替胺片;
对照品15:参照专利cn104523686a实施例1-10制得的盐酸阿考替胺片;
对照品16:参照专利cn104510719a实施例1、2、5、7制得的盐酸阿考替胺片;
对照品17:参照专利cn104510719a实施例3、4、6制得的盐酸阿考替胺片;
表6溶出度检测结果
申请人同时进行了大量的实验发现cn1063442c、wo9858918/ep0994108/cn1084739c、wo2012077673/cn103237781a/tw201229024a、wo1996036619、cn101006040b、cn102125551a、cn105924406a、cn103387552a、cn103896873a、cn104672163a、cn103665023a、cn101006040b、cn103665023a、cn103709191a、cn103709120a、cn105198832a、cn104045606a、cn200580028537、cn105439977a、cn105753810a、cn105924406a、cn101006040b、cn102040515a、cn106316979a、cn103665023a、cn1032237781a、cn1184471a、cn1261357a、cn10404560a、cn106316979a、cn102030654a现有技术制得的三水合物参照cn105769784a实施例1的处方及工艺制得盐酸阿考替胺片的30min溶出度小于70%,60min溶出度小于90%。
从上述实验结果可以看出,本发明盐酸阿考替胺水合物新晶型制得的片剂溶出度与现有技术相比,有显著提高,取得了意料不到的技术效果。
- 还没有人留言评论。精彩留言会获得点赞!