2-正丙氧基乙酰氯的制备方法与流程
本发明涉及2-正丙氧基乙酰氯化学合成领域,具体涉及2-正丙氧基乙酰氯的制备方法。
背景技术:
2-正丙氧基乙酰氯是合成氟菌唑的重要原料。1978年日本曹达公司在jps6019752专利中报道了氟菌唑产品和合成方法,但专利中只提到使用了2-正丙氧基乙酰氯原料,没有提及相关的合成方法。其它相关文献也很少报道2-正丙氧基乙酰氯的合成方法。
关于2-正丙氧基乙酸的合成,us5013864公开了以四氢呋喃为溶剂,低温条件,以氢化钠为碱,将正丙醇制备成正丙醇钠,脱溶回收四氢呋喃;再以大量的dmso存在下与得到正丙醇钠氯乙酸钠反应,得到2-正丙氧基乙酸目标产物。
《denovodesignofmultitargetligandswithaniterativefragment-growingstrategy》(journalofchemicalinformationandmodeling,erchangshang,54(4),1235-1241;2014)公开了用金属钠替代氢化钠,溴代乙酸替代氯代乙酸的方法,反应机理与us5013864专利相同,收率能达到96%,但涉及的原料成本更却加昂贵。
以上两种方法,不仅反应过程较长,涉及的原料种类也较多,操作复杂,更重要的是正丙醇钠与氯乙酸钠反应时,两种钠盐在无水环境下反应,相互接触非常困难,需要20倍量以上的非质子强极性溶剂dmso(二甲基亚砜)或者dmf(n,n-二甲基甲酰胺),产品收率低。另外反应结束后,一般需要回收dmso或者dmf,再加入酸进行酸化,然后加入有机溶剂进行萃取,脱去萃取溶剂才能得到目标产物。但因为dmso或者dmf具有高沸点、同时具有较强的酯溶性和水溶性特点,使得到2-正丙氧基乙酸产物中含有较多的dmso或者dmf。
此外,在后处理时,发现按照常规方法得到的粗品产物,含有较多的副产物2-正丙氧基乙酸丙酯,精制后产物收率低,如下式(iv)所示:
综上,在现有技术中存在正丙醇钠与氯乙酸钠接触困难,需要使用大量溶剂,且现有技术存在产品收率低、后处理纯化复杂、常出现式(iv)所示的副产物的缺陷。
技术实现要素:
本发明的目的是为了克服现有技术存在的问题,如正丙醇钠与氯乙酸钠接触困难,需要大量的dmso或者dmf溶剂,因为溶解量非常有限;而且dmso或者dmf沸点高,水溶性好,后处理复杂,产品收率低,产生副产物。本发明提供了一种2-正丙氧基乙酰氯的制备方法,该方法具有反应条件温和,不经过正丙醇钠与氯乙酸钠接触的步骤,产品收率高的优点。
为了实现上述目的,发明人通过研究发现,式(iv)所示的2-正丙氧基乙酸丙酯副产物的生成,主要是回收正丙醇溶剂时,釜中残留有正丙醇,接触到酸性物质的过程中易形成该杂质。发明人发现,在回收正丙醇后的的正丙氧基氯乙酸钠体系中,加入少量的水,溶解后再脱溶,有非常好的去除正丙醇的效果,因此,在本发明中向步骤(2)得到的产物中加入水,处理后得到的正丙氧基乙酸中,只会生成微量的2-正丙氧基乙酸丙酯,大大减少式(iv)所示的副产物,提高了产品的纯度,
2-正丙氧基乙酸制备2-正丙氧基乙酰氯的过程中,有大量的副产物氯化氢强酸生成,而在2-正丙氧基乙酸钠制备2-正丙氧基乙酸的过程中,需要较多的强酸进行酸化,此方法的原理是:先是少量的水与氯化亚砜反应,生成氯化氢,引发主反应,之后随着氯化氢的持续重复使用,完成主反应。本发明通过利用正丙基乙酸制备正丙酸乙酰氯的过程中生成的氯化氢,用于2-正丙氧基乙酸钠制备2-正丙氧基乙酸,即由酸钠直接合成酰氯,节省了氯化氢的使用。反应方程式如下,
本发明提供一种2-正丙氧基乙酰氯的制备方法,其中,该方法包括以下步骤:
(1)将氯乙酸钠和强碱在正丙醇溶剂中进行反应,得到含有式(i)所示的2-正丙氧基乙酸钠和正丙醇的混合物;
(2)将所述混合物进行蒸馏,得到除去部分正丙醇的产物;
(3)向步骤(2)得到的产物中加入水并进行蒸馏,置换出所述产物中残留的正丙醇,得到含有水和式(i)所示的2-正丙氧基乙酸钠的产物;
(4a)向步骤(3)得到的产物中加入酸,并用非水溶性溶剂进行萃取,得到式(ii)所示的2-正丙氧基乙酸,并将式(ii)所示的2-正丙氧基乙酸与酰化试剂反应,得到式(iii)所示的2-正丙氧基乙酰氯;或者,
(4b)将步骤(3)得到的产物与酰化试剂进行反应,得到式(iii)所示的2-正丙氧基乙酰氯;
在本发明中,通过将氯乙酸钠和强碱在正丙醇溶剂中进行反应,得到含有式(i)所示的2-正丙氧基乙酸钠和正丙醇的混合物,正丙醇溶剂同时作为反应试剂和反应溶剂参加反应,反应时间只需要几小时。此方法具有以下优点:首先所使用的正丙醇溶剂非常便宜,可回收,用量也只是氯乙酸钠重量的3-5倍;其次在本发明中不需要使用高活性的金属钠或者氢化钠,而是以氢氧化钠或氢氧化钾为碱,反应非常安全;最后因为正丙醇溶剂用量较少,反应时间短,同样体积反应容器产量高。
附图说明:
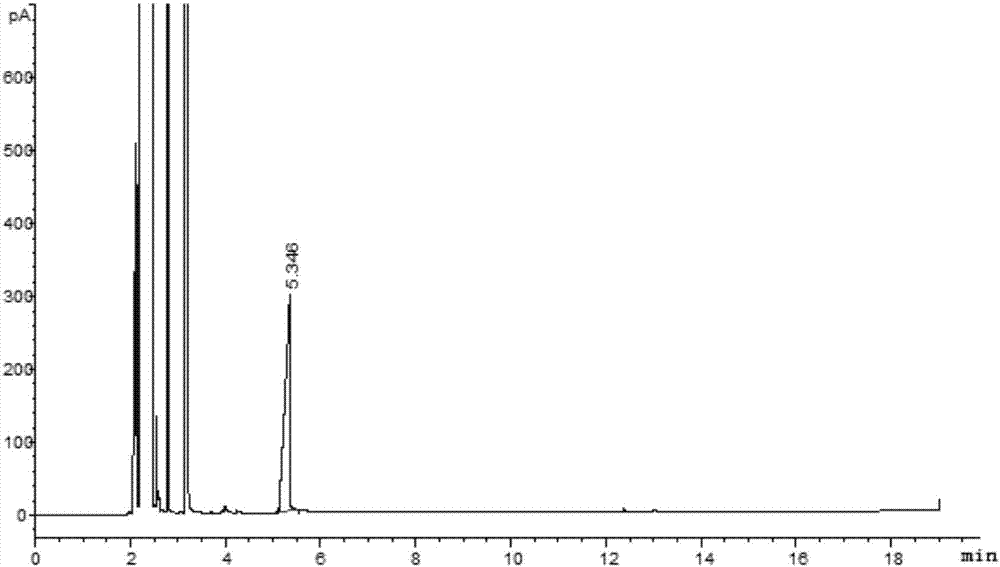
图1为本发明的实施例1的2-正丙氧基乙酸的气相色谱图;
图2为本发明的实施例1的2-正丙氧基乙酰氯的气相色谱图;
图3为对比例1的含有6.9%的2-正丙氧基乙酸丙酯副产物的2-正丙氧基乙酰氯的气相色谱图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供了一种2-正丙氧基乙酰氯的制备方法,其中,该方法包括以下步骤:
(1)将氯乙酸钠和强碱在正丙醇溶剂中进行反应,得到含有式(i)所示的2-正丙氧基乙酸钠和正丙醇的混合物;
(2)将所述混合物进行蒸馏,得到除去部分正丙醇的产物;
(3)向步骤(2)得到的产物中加入水并进行蒸馏,置换出所述产物中残留的正丙醇,得到含有水和式(i)所示的2-正丙氧基乙酸钠的产物;
(4a)向步骤(3)得到的产物中加入酸,并用非水溶性溶剂进行萃取,得到式(ii)所示的2-正丙氧基乙酸,并将式(ii)所示的2-正丙氧基乙酸与酰化试剂反应,得到式(iii)所示的2-正丙氧基乙酰氯;或者,
(4b)将步骤(3)得到的产物与酰化试剂进行反应,得到式(iii)所示的2-正丙氧基乙酰氯;
在本发明中,2-正丙氧基乙酰氯的制备方法可以为步骤(1)、(2)、(3)和(4a),即制备出式(ii)所示的2-正丙氧基乙酸,然后再制备式(iii)所示的2-正丙氧基乙酰氯;也可以为步骤(1)、(2)、(3)和(4b),不经过制备式(ii)所示的2-正丙氧基乙酸,而是直接利用制备式(iii)所示的2-正丙氧基乙酰氯,通过利用少量的水与氯化亚砜反应,生成氯化氢,引发步骤(4b)的反应,之后随着氯化氢的持续重复使用,完成步骤(4b)的反应。
在本发明中,在步骤(1)中,所述强碱可以为但不限于:氢氧化钾和/或氢氧化钠。
在本发明中,氯乙酸钠、强碱和正丙测的摩尔比以能够实现反应为目的,例如可以为1:(0.5-4.0):(1.2-15),优选为1:(0.8-1.5):(3-6)。
在本发明中,在步骤(1)中,氯乙酸钠、强碱和正丙醇溶剂的投料顺序以能够实现反应为目的。在优选的情况下,因为反应放热,具体实施方式为将正丙醇溶剂与氯乙酸钠进行充分搅拌,再加入强碱;或者,将正丙醇溶剂与强碱进行充分搅拌,再加入氯乙酸钠。
在本发明中,在步骤(1)中,所述反应的条件以能够制备含有式(i)所示的2-正丙氧基乙酸钠为目的,反应的条件可以包括但不限于:温度为20-50℃,优选为40-45℃;时间为3-8h,优选为4-5h。
在本发明中,在步骤(2)中,所述蒸馏以能够脱除正丙醇为目的,蒸馏的条件可以包括但不限于:温度为25-100℃;压力为-0.10mpa至常压,优选减压蒸馏。该压力指的是表压。
在本发明中,在步骤(3)中,以步骤(1)中1mol氯乙酸钠为基准,所述加水的量为20g-1000g,优选为50g-700g,更优选为100g-450g。当采用(4a)方法,因为置换出正丙醇后,还需要水来溶解氯化钠和产物,所以用水量优选为400g-450g。当采用(4b)方法,不需要留存水来溶解氯化钠和产物,所以用水量可以减少,优选为100g-150g。
在本发明中,在步骤(3)中所述蒸馏的条件以能够置换出步骤(2)产物中残留的正丙醇为目的,蒸馏的条件以能够除去大部分的正丙醇为目的,蒸馏的条件可以包括但不限于:温度为25-100℃;压力为-0.01mpa至常压,优选减压蒸馏。该压力指的是表压。
在本发明中,在步骤(4a)中,所述酸可以为本领域常规的酸性超过2-正丙氧基乙酸的无机酸或者有机酸,例如可以为盐酸、硫酸或磷酸、二氯乙酸,优选为盐酸。
在本发明中,在步骤(4a)中,所述酸的加入量,以式(i)所示化合物全部转化为式(ii)所示化合物为目的,调节ph值为1-2,也就是说,加入酸的量,使得溶液体系的ph值为1-2,然后再用非水溶性溶剂进行萃取。
在本发明中,在步骤(4a)中,所述非水溶性溶剂为乙酸丙酯、乙酸异丙酯、乙酸乙酯、乙酸丁酯、二氯甲烷、二氯乙烷、氯苯、甲苯和二甲苯中的一种或多种;优选为乙酸乙酯或乙酸丙酯。
在本发明中,在步骤(4a)和(4b)中,所述酰化试剂为氯化亚砜、草酰氯、磺酰氯、光气、三光气、三氯化磷和三氯氧磷中的一种或多种;优选为氯化亚砜或三氯化磷。
在本发明中,在步骤(4a)中,所述酰化试剂与式(ii)所示的2-正丙氧基乙酸的摩尔比以能够制备式(iii)所示的2-正丙氧基乙酰氯为目的,例如可以为(1-5):(1-3),优选为(1.2-1.5):1。
在本发明中,在步骤(4b)中,所述酰化试剂与式(i)所示2-正丙氧基乙酸钠的摩尔比以能够制备式(iii)所示的2-正丙氧基乙酰氯为目的,例如可以为(1-5):(1-3),优选为(1-2):1。
以下将通过实施例对本发明进行详细描述。
在以下实施例中,物质的含量通过aglient公司生产,型号为6890gc的气相色谱进行分析,该气相色谱配置氢离子火焰检测器,具体分析方法见表1。
表1
实施例1
(1)制备式(i)所示的2-正丙氧基乙酸钠
在1000ml反应瓶中,加入1.0mol氯乙酸钠(浓度为95%,122.6g)和4mol正丙醇,搅拌均匀后,加入1.3mol氢氧化钠(98%,53.1g),升温到45℃,保温5小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在-0.05mpa压力下,85℃下进行减压脱溶回收大部分正丙醇,得到除去部分正丙醇的产物。
向产物中加入450g的水并在-0.05mpa下,100℃下进行减压蒸馏,置换出所述产物中残留的醇,得到含有式(i)所示的2-正丙氧基乙酸钠的水溶液,
(2)制备式(ii)所示的2-正丙氧基乙酸
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,用30%的盐酸调节ph值为1,每次用400ml乙酸乙酯进行萃取,萃取三次,合并萃取液,减压脱去乙酸乙酯,然后进行气相色谱检测,谱图如附图1所示,时间在5.346min的峰为式(ii)所示的2-正丙氧基乙酸,
(3)制备式(iii)所示的2-正丙氧基乙酰氯
向1mol式(ii)所示的2-正丙氧基乙酸中滴加180.3g氯化亚砜(浓度为99%,1.5mol),滴加结束后,缓慢升温到回流,保温反应1小时,取样分析,直至2-正丙氧基乙酸的含量小于0.5%,进行气相色谱检测,谱图如图2所示,时间在4.124min的峰为式(iii)所示的2-正丙氧基乙酰氯的粗品,其中,式(iv)所示的2-正丙氧基乙酸丙酯副产物的含量为0.07%。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到提纯的式(iii)所示的2-正丙氧基乙酰氯,重量为120.6g,收率为88.3%。
实施例2
(1)制备式(i)所示的2-正丙氧基乙酸钠
在1000ml反应瓶中,加入1.0mol氯乙酸钠(95%,122.6g)和6mol正丙醇,搅拌均匀后,加入1.5mol氢氧化钾(98%,85.9g),升温到40℃,保温4小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在-0.1mpa压力下,50℃下进行减压脱溶回收大部分正丙醇,得到除去部分正丙醇的产物。
向产物中加入100g的水并在-0.1mpa下,50℃下进行减压蒸馏,置换出所述产物中残留的醇,得到含有式(i)所示的2-正丙氧基乙酸钠的水溶液,
(2)制备式(iii)所示的2-正丙氧基乙酰氯
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,向1mol式(i)所示的2-正丙氧基乙酸钠中滴加入240.4g氯化亚砜(99%,2mol),滴加结束后,缓慢升温到回流,回流反应1小时,取样分析,直至2-正丙氧基乙酸的浓度小于0.5%,按照实施例1的方法检测得到式(iii)所示的2-正丙氧基乙酰氯的粗品,以及含量为0.08%的式(iv)所示的2-正丙氧基乙酸丙酯副产物。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到提纯的式(iii)所示的2-正丙氧基乙酰氯,重量为116.2g,收率85.1%。
实施例3
(1)制备式(i)所示的2-正丙氧基乙酸钠
在1000ml反应瓶中,加入1.0mol氯乙酸钠(95%,122.6g)和3mol正丙醇,搅拌均匀后,加入0.8mol氢氧化钠(98%,32.7g),升温到50℃,保温3小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在-0.1mpa压力下,50℃下进行减压脱溶回收大部分正丙醇,得到除去部分正丙醇的产物。
向产物中加入700g的水并在常压、100℃下进行蒸馏,置换出所述产物中残留的醇,得到含有式(i)所示的2-正丙氧基乙酸钠的水溶液,
(2)制备式(ii)所示的2-正丙氧基乙酸
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,用30%的磷酸调节ph值为2,每次用400ml乙酸异丙酯进行萃取,萃取三次,合并萃取液,减压脱去乙酸异丙酯,按照实施例1的方法进行气相色谱检测,说明得到了式(ii)所示的2-正丙氧基乙酸,
(3)制备式(iii)所示的2-正丙氧基乙酰氯
向1mol式(ii)所示的2-正丙氧基乙酸中滴加166.5g三氯化磷(99%,1.2mol),滴加结束后,缓慢升温到回流,保温反应1小时,取样分析,直至2-正丙氧基乙酸的含量小于0.5%,按照实施例1的方法检测得到式(iii)所示的2-正丙氧基乙酰氯的粗品,以及含量为0.01%的式(iv)所示的2-正丙氧基乙酸丙酯副产物。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到提纯的式(iii)所示的2-正丙氧基乙酰氯,重量为120.2g,收率为88.0%。
实施例4
(1)制备式(i)所示的2-正丙氧基乙酸钠
在1000ml反应瓶中,加入1.0mol氯乙酸钠(95%,122.6g)和15mol正丙醇,搅拌均匀后,加入1.3mol氢氧化钾(98%,74.4g),升温到20℃,保温8小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在100℃下进行常压脱溶回收大部分正丙醇,得到除去部分正丙醇的产物。
向产物中加入50g的水并在-0.09mpa,50℃下进行减压蒸馏,置换出所述产物中残留的醇,得到含有式(i)所示的2-正丙氧基乙酸钠的水溶液,
(2)制备式(ii)所示的2-正丙氧基乙酰氯
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,向1mol式(i)所示的2-正丙氧基乙酸钠中滴加入136.3g磺酰氯(99%,1mol),滴加结束后,缓慢升温到回流,回流反应1小时,取样分析,直至2-正丙氧基乙酸的浓度小于0.5%,按照实施例1的方法检测得到式(iii)所示的2-正丙氧基乙酰氯的粗品,以及含量为0.09%的式(iv)所示的2-正丙氧基乙酸丙酯副产物。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到式(iii)所示的2-正丙氧基乙酰氯,重量为110g,以氯乙酸钠计,收率84.5%。
对比例1
(1)制备式(i)所示的2-正丙氧基乙酸钠
在1000ml反应瓶中,加入1.0mol氯乙酸钠(浓度为95%,122.6g)和4mol正丙醇,搅拌均匀后,加入1.3mol氢氧化钠(98%,53.1g),升温到45℃,保温5小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在-0.05mpa压力下,85℃下进行减压脱溶回收大部分正丙醇,得到含有式(i)所示的2-正丙氧基乙酸钠的产物,
(2)制备式(ii)所示的2-正丙氧基乙酸
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,用30%的盐酸调节ph值为1,每次用400ml乙酸乙酯进行萃取,萃取三次,合并萃取液,减压脱去乙酸乙酯,按照实施例1的方法进行气相色谱检测,说明得到了式(ii)所示的2-正丙氧基乙酸,
(3)制备式(iii)所示的2-正丙氧基乙酰氯
向1mol式(ii)所示的2-正丙氧基乙酸中滴加180.3g氯化亚砜(浓度为99%,1.5mol),滴加结束后,缓慢升温到回流,保温反应1小时,取样分析,直至2-正丙氧基乙酸的含量小于0.5%,按照实施例1的方法检测得到式(iii)所示的2-正丙氧基乙酰氯的粗品,以及含量为6.9%的式(iv)所示的2-正丙氧基乙酸丙酯副产物,如图3所示,时间在4.1min的峰为式(iii)所示的2-正丙氧基乙酰氯的粗品,时间在6.092min的峰为2-正丙基乙酸丙酯含量,杂质峰较明显。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到式(iii)所示的2-正丙氧基乙酰氯,重量为104.7g,收率为77.0%。
通过对比实例1与对比例1的结果可以看出,除去部分正丙醇的产物中不加入水,2-正丙氧基乙酸丙酯的杂质明显较多,导致2-正丙氧基乙酰氯收率的降低。
对比例2
在1000ml反应瓶中,加入1.0mol氯乙酸钠(浓度为95%,122.6g)和2.0mol正丙醇,搅拌均匀后,加入0.35mol氢氧化钠(98%,14.3g),升温到45℃,保温5小时,取样分析,氯乙酸钠的转化率达到99.5%后停止反应。
在-0.05mpa压力下,85℃下进行减压脱溶回收大部分正丙醇,得到除去部分正丙醇的产物。
向产物中加入450g的水并在-0.05mpa下,100℃下进行减压蒸馏,置换出所述产物中残留的醇,得到含有式(i)所示的2-正丙氧基乙酸钠的水溶液,
(2)制备式(ii)所示的2-正丙氧基乙酸
将得到产物的反应瓶与冷凝器和尾气吸收装置进行连通,用30%的盐酸调节ph值为1,每次用400ml乙酸乙酯进行萃取,萃取三次,合并萃取液,减压脱去乙酸乙酯,按照实施例1的方法进行气相色谱检测,说明得到了式(ii)所示的2-正丙氧基乙酸,
(3)制备式(iii)所示的2-正丙氧基乙酰氯
向1mol式(ii)所示的2-正丙氧基乙酸中滴加180.3g氯化亚砜(浓度为99%,1.5mol),滴加结束后,缓慢升温到回流,保温反应1小时,取样分析,直至2-正丙氧基乙酸的含量小于0.5%。按照实施例1的方法检测得到式(iii)所示的2-正丙氧基乙酰氯的粗品,以及含量为0.05%的式(iv)所示的2-正丙氧基乙酸丙酯副产物。
将反应液转移到带精馏的反应器进行精制,收集-0.09mpa,90-95℃馏分产物,得到式(iii)所示的2-正丙氧基乙酰氯,重量为79.3g,收率为58.3%,
通过对比实例1与对比例2的结果可以看出,降低氢氧使钠和正丙醇的用量,2-正丙氧基乙酰氯的收率下降。
对比例3
在2000ml反应瓶中,加入200ml四氢呋喃,36.8g正丙醇,用冰盐水降温到0℃,加入24g的60%的氢化钠,升到常温,反应1小时,减压回收四氢呋喃,加入1000ml的dmso和49.1g氯代乙酸钠(95%,0.4mol),常温反应过夜,减压回收dmso,加入300ml水,用30%盐酸中和到ph为1,用800ml乙酸乙酯分三次萃取,合并有机相,脱溶得到式(i)所示的2-正丙氧基乙酰氯,质量为55.1.g,含量为76.5%,(其中dmso含量22%),收率为77.5%。
通过实施例和对比例的结果可以看出,通过加水精馏的方法去除正丙醇的方法,副产物2-正丙氧基乙酸丙酯从6.9%减少至0.1%以下,提高了2-正丙氧基乙酰氯的收率。
采用本发明方法制备的2-正丙氧基乙酰氯,不经过正丙醇钠与氯乙酸钠接触的步骤,溶剂用量少,而且能够提高产品的收率,收率能够从77.5%提高至84.5%以上,甚至88.3%。
以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种制备2,3,4‑三乙酰基‑1‑(2‑氯‑4‑硝基‑苯基)‑alpha‑L岩藻吡喃糖苷的方法与制造工艺
- 利用混酚制备有机磷配体并进一步制备己二腈的方法与制造工艺
- 一种左西孟旦药物中间体对乙酰氨基苯丙酮的合成方法与制造工艺
- 一种柳氨酚药物中间体N‑对羟基苯基水杨酰胺的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 含有高剂量和低剂量药物的组合的口腔崩解片组合物的制造方法与工艺
- 一种新型甲氨基阿维菌素水分散粒剂及其制备方法与制造工艺
- 一种药物中间体邻乙酰氨基酚的合成方法与制造工艺
- 一种3‑氨基邻苯二甲酸二甲酯的制备方法与制造工艺
- 三芳胺端基含量可调的主链含S,S‑二氧‑二苯并噻吩的聚合物及其制备方法与应用与制造工艺
- 一种制备5-乙酰乙酰氨基苯并咪唑酮工艺过程中大颗粒物分离装置的制造方法
- 一种用于回收5-乙酰乙酰氨基苯并咪唑酮粉末的装置的制造方法
- 一种用于制备5-乙酰乙酰氨基苯并咪唑酮的氯苯与水分离装置的制造方法
- 对乙酰氨基酚的合成工艺的制作方法
- 聚谷氨酸修饰电极及其在检测药物对乙酰氨基酚含量中的应用
- 一种地西泮药物中间体2-氯乙酰氨基-5-氯二苯酮的合成方法
- 一种氨基甲酰尿嘧啶衍生物及其应用
- 2-乙酰氨基-1,5,-脱水-2-脱氧-d-葡萄糖醇在制备抗水稻白叶枯病菌活性药物中的应用
- 对乙酰氨基酚精制工段所产生的废活性炭的再利用工艺的制作方法
- 使用其中进行了调控使赖氨酰-tRNA合成酶被表达或不被表达的细胞或球状聚集的细胞 ...的制作方法
- 一种制备2,3,4‑三乙酰基‑1‑(2‑氯‑4‑硝基‑苯基)‑alpha‑L岩藻吡喃糖苷的方法与制造工艺
- 利用混酚制备有机磷配体并进一步制备己二腈的方法与制造工艺
- 一种左西孟旦药物中间体对乙酰氨基苯丙酮的合成方法与制造工艺
- 一种柳氨酚药物中间体N‑对羟基苯基水杨酰胺的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 含有高剂量和低剂量药物的组合的口腔崩解片组合物的制造方法与工艺
- 一种新型甲氨基阿维菌素水分散粒剂及其制备方法与制造工艺
- 一种药物中间体邻乙酰氨基酚的合成方法与制造工艺
- 一种3‑氨基邻苯二甲酸二甲酯的制备方法与制造工艺
- 三芳胺端基含量可调的主链含S,S‑二氧‑二苯并噻吩的聚合物及其制备方法与应用与制造工艺