纯化蛋白质的方法与流程
本申请要求2016年8月12日提交的美国临时申请no.62/374,337的权益,将其内容通过提述明确并入本文。
发明背景
单克隆抗体(mab)及其衍生产物(例如fc融合蛋白)在治疗一些最具挑战性的人类疾病中起重要作用,原因在于这些类型的生物制剂的安全性、功效和高质量[1]。单克隆抗体市场正在显著快速增长,据估计,到2020年单克隆抗体产品的全球总销售额将达到约1250亿美元[2]。因此,强大的商业色谱纯化方法的开发对患者至关重要[3]。纯化方法通常使用蛋白a色谱法进行捕获,然后进行一个或两个精制步骤[4]。对于那些具有碱性等电点(pi)的mab和fc融合蛋白,阳离子交换色谱(cex)传统上被认为是精制步骤的首选,主要原因在于相对简单的色谱柱行为、产物相关杂质(例如,hmw)的有效去除、以及静电相互作用通常对蛋白质结构的轻微影响[5,6]。相比之下,在疏水相互作用色谱(hic)介质的表面上吸附后,蛋白质通常经历部分解折叠[7-9]。
近年来,越来越多的案例报道了在cex中的相当反常规的蛋白质结合和洗脱行为。例如,voitl等人[10,11]报道了在fractogelemdsehicap上的人血清白蛋白的线性盐梯度期间的双峰洗脱曲线。由于在两个洗脱峰中没有观察到hmw增加,因此假设该蛋白质以两种不同的构象与树脂结合,需要不同的盐浓度将其完全洗脱。gillespie等人[12]报道了使用线性盐梯度在几种cex树脂中的不稳定的非糖基化igg1的双峰洗脱图,并且晚期洗脱峰含有比早期洗脱峰更多的聚集物。氢氘交换和傅立叶变换红外光谱(ftir)结果表明,第二峰起源于树脂诱导的抗体变性,其可通过使用优先排除的溶质(比如精氨酸)而减轻[13-15]。这项工作评估了许多与cex操作相关的因素,但没有详细论述聚集机制。guo和carta[16-18]报道了糖基化igg2的双峰cex洗脱行为。这种效应对于聚合物功能化树脂fractogelemdso3-来说是显著的,但对于没有接枝聚合物的大孔树脂(例如unosphererapids)几乎不存在,从而导致这样的假设,即,蛋白质通过触手型(tentacle)聚合物扩散使蛋白质结构不稳定并引起聚集物形成。应该注意的是,仅当结合的蛋白质被保持延长时段时才观察到双峰洗脱行为。luo等人[19]报道了igg2在盐洗脱后的双峰洗脱行为。这项工作表明,igg2可以在高盐和高蛋白质浓度下形成可逆自缔合(rsa),并且rsa物质可以比单体更强地与树脂结合,从而导致峰分裂。在另一项研究中,luo等人[20]报道了igg4的分裂峰现象,并将其与基于组氨酸-质子化的电荷变体的分离联系起来。
生物治疗剂的多样化柱行为最终可归因于其复杂的分子特性。例如,尽管mab和fc融合蛋白共有相似的可结晶片段(fc)区域,但由于高度可变的互补决定区(cdr)和fc区中的糖基化,它们的溶液性质可能显著不同[21]。据报道,一些mab在低ph(ph2-4)和高盐浓度下易于变性和聚集[22]。buchner等人[23]报道,免疫球蛋白可在低ph(<3)下形成“a-状态”,其特征在于高度二级结构,具有增加的疏水性以及在高盐中的缓慢聚集趋势。latypov等人[24,25]对人igg1和igg2的酸诱导的解折叠和聚集进行了广泛研究,所述酸诱导的解折叠和聚集主要取决于位于fc区的ch2结构域的稳定性。取决于溶液条件,诸如mab的蛋白质可以部分地解折叠并且彼此形成不可逆的聚集物或可逆的簇,同时保持它们的天然结构[26,27]。特别是在高蛋白质浓度下,可逆或不可逆的mab缔合通常由蛋白质-蛋白质吸引力驱动,所述吸引力主要由于在mab表面上的异种电荷分布所致[28-30],而最近关于在电解质存在下二聚体igg1mab微结构形成的报道也表明重要的非静电贡献,比如疏水相互作用[26]。由于缺乏分子间和分子内结构域稳定性,与完整mab相比,fc融合蛋白甚至更可能经历构象变化。fast等人[31]报道,在40℃当ph值从7.5降至6.0时,fc融合蛋白阿巴西普(abatacept)(orencia)快速聚集。构象变化和聚集物形成归因于cdr(ctla-4)和ch2结构域的不稳定性,所述结构域解折叠而形成易于聚集的熔融球状结构。
此外,由于移动相和固定相之间复杂的离子平衡,蛋白质在cex柱中暴露的溶液条件可能难以确定。因此,在理解复杂的色谱柱现象时,可能会出现蛋白质溶液性质的影响与色谱过程的影响相混淆的情况。例如,据报道,由于缓冲盐离子和h+/oh-离子之间的竞争平衡,在盐洗脱期间在cex中可能发生意外的ph下降[32-34]。当在洗脱步骤中引入高盐缓冲液时,流动相中的阳离子置换固定相中的h+离子。然后这些释放的h+离子进入流动相并引起洗脱物中的ph暂时降低。除了洗脱后的高孔内蛋白浓度外,对于不稳定的蛋白质还应额外注意,在高盐和低ph的局部环境情况的这种瞬时条件下可引起蛋白质变性和聚集。
因此,本领域需要改进的蛋白质纯化方法,其可用于减少蛋白质纯化过程中的聚集形成。
发明概述
在某些实施方案中,本发明提供了在阳离子交换(cex)色谱中以降低的聚集形成水平纯化感兴趣的蛋白质的方法,所述方法包括:(a)提供包含感兴趣的蛋白质和一种或多种污染物的混合物;(b)将混合物上样到偶联有精氨酸的cex树脂上;和(c)从树脂中洗脱感兴趣的蛋白质,从而在cex色谱中以降低的聚集形成水平纯化感兴趣的蛋白质。例如,混合物包含澄清原液(clarifiedbulk)或细胞培养上清液(例如来自哺乳动物细胞、细菌细胞或真菌细胞培养物的上清液)。在一个具体实例中,混合物是来自中国仓鼠卵巢(cho)细胞培养物的上清液。为了说明,污染物选自宿主细胞蛋白、宿主细胞代谢物、宿主细胞组成蛋白、核酸、内毒素、病毒、产物相关污染物、脂质、培养基添加剂和培养基衍生物。为了说明,感兴趣的蛋白质选自抗体(例如,选自人抗体、人源化抗体和嵌合抗体的单克隆抗体)、抗体片段和fc融合物。
在某些方面,本发明的cex树脂的基础基质材料选自琼脂糖、纤维素、葡聚糖、壳聚糖、聚(甲基丙烯酸酯)、丙烯酸聚合物和聚(苯乙烯-二乙烯基-苯)。使用选自磺酸盐、羧酸、羧甲基磺酸、磺基异丁基、磺乙基、羧基、磺丙基、磺酰基、磺酰氧乙基(sulphoxyethyl)和正磷酸盐的阳离子交换配体功能性制备cex树脂。在具体实施方案中,偶联有精氨酸的cex树脂是精氨酸-磺丙基(arg-sp)树脂(例如,精氨酸-磺丙基(arg-sp)琼脂糖树脂)。
在某些方面,在cex色谱步骤之前,通过选自蛋白a亲和色谱法和蛋白g亲和色谱法的亲和色谱法制备混合物。任选地,亲和色谱法是蛋白a亲和色谱法。在某些方面,本发明的方法进一步包括一种或多种另外的色谱基质,比如阴离子交换色谱、疏水相互作用色谱和/或混合模式色谱。
在其他实施方案中,本发明提供了偶联有精氨酸的阳离子交换(cex)树脂。例如,cex树脂的基础基质材料选自琼脂糖、纤维素、葡聚糖、壳聚糖、聚(甲基丙烯酸酯)、丙烯酸聚合物和聚(苯乙烯-二乙烯基-苯)。例如,使用选自磺酸盐、羧酸、羧甲基磺酸、磺基异丁基、磺乙基、羧基、磺丙基、磺酰基、磺酰氧乙基(sulphoxyethyl)和正磷酸盐的阳离子交换配体功能性制备cex树脂。在具体实施方案中,偶联有精氨酸的cex树脂是精氨酸-磺丙基(arg-sp)树脂,比如精氨酸-磺丙基(arg-sp)琼脂糖树脂。
附图简述
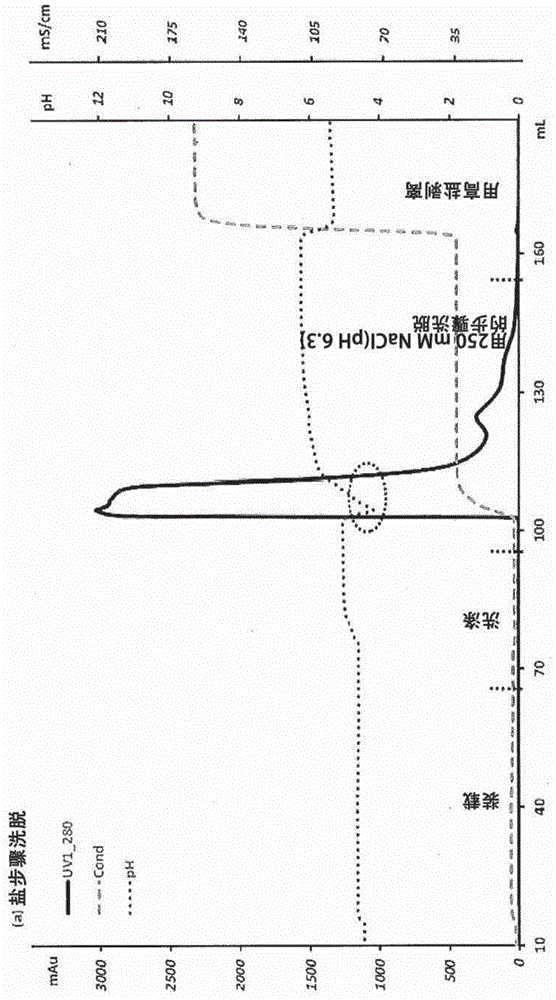
图1a显示了使用盐步骤洗脱的spsff的色谱图。在ph4.5以30g/l树脂装填cex柱,用50mm乙酸钠在ph5.0洗涤,分别用50mm乙酸钠、250mmnacl在ph6.3(a)或用50mm乙酸钠在ph8.3(b)洗脱。
图1b显示了使用ph步骤洗脱的spsff的色谱图。在ph4.5以30g/l树脂装填cex柱,用50mm乙酸钠在ph5.0洗涤,分别用50mm乙酸钠、250mmnacl在ph6.3(a)或用50mm乙酸钠在ph8.3(b)洗脱。
图2显示了盐步骤和ph步骤洗脱物(在spsff上)、装载材料(在50mm乙酸钠,ph4.5缓冲液中)和在50mm乙酸钠、250mmnacl、ph4.5缓冲液中的对照溶液样品的sec谱。
图3显示了所提出的聚集和解离过程的机制。
图4显示了对spsff的洗脱物聚集物水平的柱装载效应。在ph步骤洗脱(t=0天)和在4℃温育5天(t=5天)后立即进行sec测试。
图5显示了天然单链和游离单链的sap评分。红色和蓝色分别代表疏水和亲水表面区。
图6a显示了ph步骤洗脱物的聚集物水平的时间依赖性。在ph4.5以30g/l树脂装载所有柱。
图6b显示了不同树脂的聚集可逆性的时间依赖性。在ph4.5以30g/l树脂装载所有柱。
图7显示了arg-sp混合模式琼脂糖珠的示意图。
图8显示了使用不同树脂的ph步骤洗脱物的聚集物水平。在ph4.5以10g/l树脂装载cex柱,并用50mm乙酸钠、ph8.3缓冲液洗脱。
图9显示了在不同时间点的spsff洗脱池(在ph4.5以30g/l树脂装载)的sec。洗脱后,在4℃温育0、2、4、6、8、24和120小时之后测定ph步骤洗脱物。装载材料的聚集物水平为1.5%。
图10显示了通过计算表面上的静电势进行的不同ph下的电荷建模;表面上的负电荷片区(negativepatch)随着ph的增加而增加。
图11显示了与每个残基的ph4下的值相比计算的蛋白质电荷电位变化。
图12显示了不同ph条件下spsff、cmsff和结合蛋白的dsf结果。
图13显示了当与spsff结合时在cex装载ph、洗脱物聚集物水平、蛋白质净电荷和蛋白质熔化温度(tm)之间的相关性。
图14显示了syproorange染料实验,其显示了树脂的疏水性。将树脂与syproorange染料一起在25mm乙酸钠(ph5)中温育。将所有样品在辊式混合器上以180rpm倒转5分钟,然后在拍照之前使其经历重力沉降。实验条件参见表4。
发明详述
在某些实施方案中,本发明提供了在阳离子交换(cex)色谱中以降低的聚集形成水平纯化感兴趣的蛋白质的方法,所述方法包括:(a)提供包含感兴趣的蛋白质和一种或多种污染物的混合物;(b)将混合物上样到偶联有精氨酸的cex树脂上;和(c)从树脂中洗脱感兴趣的蛋白质,从而在cex色谱中以降低的聚集形成水平纯化感兴趣的蛋白质。在具体实施方案中,偶联有精氨酸的cex树脂是精氨酸-磺丙基(arg-sp)树脂,比如精氨酸-磺丙基(arg-sp)琼脂糖树脂。例如,混合物包含澄清原液(clarifiedbulk)或细胞培养上清液。为了说明,感兴趣的蛋白质选自抗体、抗体片段和fc融合物。任选地,在cex色谱步骤之前,通过亲和色谱法(例如蛋白a亲和色谱法和蛋白g亲和色谱法)制备混合物。任选地,本发明的方法进一步包括一种或多种另外的色谱基质,比如阴离子交换色谱、疏水相互作用色谱和/或混合模式色谱。
在其他实施方案中,本发明提供了偶联有精氨酸的阳离子交换(cex)树脂。在具体实施方案中,偶联有精氨酸的cex树脂是精氨酸-磺丙基(arg-sp)树脂,比如精氨酸-磺丙基(arg-sp)琼脂糖树脂。
定义
如本文中使用的,术语“纯化”和“分离”可互换使用,并且是指从含有感兴趣的蛋白质(例如抗体)的混合物中除去污染物。
如本文中使用的,在其最广泛的意义上使用术语“感兴趣的蛋白质”,其包括存在于需要纯化的混合物中的任何蛋白质(天然的或重组的)。这样的感兴趣的蛋白质包括但不限于激素、生长因子、细胞因子、免疫球蛋白(例如抗体)、含免疫球蛋白样结构域的分子(例如含锚蛋白或纤连蛋白结构域的分子)和fc融合蛋白。如本文中使用的,术语“fc融合蛋白”意味着涵盖治疗性蛋白质,其包含免疫球蛋白衍生模块(即fc模块)和衍生自第二种非免疫球蛋白蛋白质的模块。
如本文中使用的,“混合物”包含感兴趣的蛋白质(需要纯化)和一种或多种污染物(即杂质)。在一个实施方案中,混合物由表达感兴趣的蛋白质的宿主细胞或生物产生(天然或重组)。这样的混合物包括,例如,细胞培养物、细胞裂解物和澄清原液(例如,澄清的细胞培养上清液)。
如本文中使用的,在其最广泛的意义上使用术语“污染物”,以覆盖混合物内任何不期望的组分或化合物。在细胞培养物、细胞裂解物或澄清原液(例如,细胞培养上清液)中,污染物包括例如存在于细胞培养基中的宿主细胞核酸(例如dna)和宿主细胞蛋白、与感兴趣的蛋白质相关或从其衍生的蛋白质(例如,蛋白水解片段)以及其他的产物相关污染物(例如,感兴趣的蛋白质的截短和聚集形式)。宿主细胞污染蛋白包括但不限于由宿主细胞以天然或重组方式产生的蛋白质。
如本文中使用的,“洗涤”是指使适当的缓冲剂穿过或通过阳离子交换树脂。
如本文中使用的,“洗脱”是指从阳离子交换树脂中除去感兴趣的蛋白质(例如抗体),其通过改变阳离子交换树脂周围的缓冲液的ph和/或离子强度使得缓冲液与所述分子竞争离子交换材料上的带电位点来实现。
如本文中使用的,“细胞培养物”是指在液体培养基中的产生感兴趣的蛋白质的细胞。细胞可以来自任何生物,包括例如细菌、真菌、哺乳动物或植物。适合的液体培养基包括例如营养培养基和非营养培养基。
如本文中使用的,术语“澄清原液”是指基本上除去颗粒物质(例如,细胞)的混合物。澄清原液包括通过例如过滤或离心从中基本上除去细胞或细胞碎片的细胞培养上清液或细胞裂解物。
在最广泛的意义上使用术语“抗体”,以涵盖任何已知抗体类型,包括但不限于,单克隆抗体(包括全长单克隆抗体)、多克隆抗体、单特异性抗体、多特异性抗体(如双特异性抗体)、免疫粘附素、抗体-免疫粘附素嵌合体;人源化抗体、人抗体、嵌合抗体、单链抗体、合成抗体、重组抗体、杂交抗体、突变抗体、移植抗体或体外生成的抗体。抗体可以是全长抗体或抗体片段。抗体可选自任何已知的抗体同种型,例如iga、igg、igd、ige、igm。抗体可以是单体、二聚体或多聚体(例如,三聚体或五聚体)。
“抗体片段”包括全长抗体的至少一部分,并且通常是其抗原结合区或可变区。抗体片段的实例包括fab、fab'、f(ab')2和fv片段;单链抗体分子;双抗体;线性抗体;和由工程化抗体片段形成的多特异性抗体。
在传统意义上使用术语“单克隆抗体”,其是指从基本上同质的抗体群获得的抗体,使得除了可能以少量存在的可能的天然存在的突变之外,构成该群的单独的抗体是完全相同的。单克隆抗体针对单个抗原位点是高度特异性的。这与通常包括针对抗原的不同决定簇(表位)的不同抗体的多克隆抗体制剂相反,而单克隆抗体是针对抗原上的单一决定簇。当描述抗体时,术语“单克隆的”表示从基本上同质的抗体群获得的抗体的特性,并且不被解释为需要通过任何特定的方法产生抗体。例如,在本发明中使用的单克隆抗体可以通过首先由kohler等人在nature256:495(1975)中描述的常规杂交瘤技术来产生,或者可以使用重组dna方法(参见例如美国专利no.4,816,567)来制造它们。也可以从噬菌体抗体文库中分离单克隆抗体,例如,使用clackson等人在nature352:624-628(1991);marks等人,j.mol.biol.222:581-597(1991);和美国专利no.5,223,409;5,403,484;5,571,698;5,427,908;5,580,717;5,969,108;6,172,197;5,885,793;6,521,404;6,544,731;6,555,313;6,582,915;和6,593,081中描述的技术。
本文所述的单克隆抗体包括这样的“嵌合”和“人源化”抗体:其中重链和/或轻链的一部分与衍生自特定物种或属于特定抗体类别或亚类的抗体中的相应序列相同或同源,而所述链的其余部分与衍生自另一物种或属于另一种抗体类别或亚类的抗体中的相应序列相同或同源;还包括这样的抗体的片段,只要它们表现出期望的生物活性即可(美国专利no.4,816,567;和morrison等人,proc.natl.acad.sci.usa,81:6851-6855(1984)。非人(例如,鼠)抗体的“人源化”形式是含有衍生自非人免疫球蛋白的最小序列的嵌合抗体。大多数情况下,人源化抗体是人免疫球蛋白(受体抗体),其中受体的高变区残基被来自具有期望的特异性、亲和力和能力的诸如小鼠、大鼠、兔或非人灵长类动物等非人物种(供体抗体)的高变区残基替换。在一些情况下,人免疫球蛋白的fv框架区(fr)残基被相应的非人残基替换。此外,人源化抗体可以包含在受体抗体中或在供体抗体中未发现的残基。进行这些修饰以进一步改善抗体性能。一般而言,人源化抗体将包含至少一个(通常两个)可变结构域中的基本上所有的可变结构域,其中所有或基本上所有的高变环对应于非人免疫球蛋白的高变环,并且所有或基本上所有的fr区是人免疫球蛋白序列的fr区。人源化抗体任选地还将包含免疫球蛋白恒定区(fc)(一般是人免疫球蛋白的恒定区)的至少一部分。关于更多详情,参见jones等人,nature321:522-525(1986);riechmann等人,nature332:323-329(1988);和presta,curr.op.struct.biol.2:593-596(1992)。
基于如上文所述制备的鼠单克隆抗体的序列,可以制备嵌合抗体或人源化抗体。可以从感兴趣的鼠杂交瘤获得编码重链和轻链免疫球蛋白的dna,并使用标准分子生物学技术将其工程改造为含有非鼠(例如,人)免疫球蛋白序列。例如,为了产生嵌合抗体,可使用本领域已知的方法将鼠可变区与人恒定区连接(例如,参见授予cabilly等人的美国专利no.4,816,567)。为了产生人源化抗体,可使用本领域已知的方法将鼠cdr区插入人框架中(例如,参见授予winter的美国专利no.5,225,539以及授予queen等人的美国专利no.5,530,101;5,585,089;5,693,762和6,180,370)。
本文所述的单克隆抗体还包括“人”抗体,其可以从各种来源分离,包括例如来自人类患者的血液,或使用转基因动物重组制备。这样的转基因动物的实例包括km-
如本文中使用的,术语“色谱法”是指混合物中感兴趣的溶质(例如感兴趣的蛋白质)与混合物中的其他溶质通过吸附剂渗滤分离的过程,所述吸附剂在该过程的特定缓冲条件下由于溶质的性质(比如pi、疏水性、大小和结构)而强弱不同地吸附或保留溶质。
术语“离子交换”和“离子交换色谱法”是指其中感兴趣的可电离溶质(例如,混合物中的感兴趣的蛋白质)在适当的ph和导电性条件下与连接(例如,通过共价连接)至固相离子交换材料的带相反电荷的配体相互作用的色谱过程,使得感兴趣的溶质与带电化合物非特异性地相互作用,所述相互作用比与混合物中的溶质杂质或污染物的相互作用更多或更少。混合物中的污染性溶质可以比感兴趣的溶质更快或更慢地从离子交换材料柱中洗涤,或与树脂结合或从其排除。“离子交换色谱法”具体包括阳离子交换(cex)、阴离子交换和混合模式色谱法。
“阳离子交换树脂”或“cex树脂”是指带负电荷的固相,并且它具有与穿过或通过该固相的水溶液中的阳离子交换的游离阳离子。可以使用与适于形成阳离子交换树脂的固相附接的任何带负电荷的配体,例如羧酸盐、磺酸盐。可商购的阳离子交换树脂包括但不限于例如具有下列基团的那些树脂:基于磺酸盐的基团(例如,来自gehealthcare的monos、minis、source15s和30s、spsepharosefastflowtm、spsepharosehighperformance,来自tosoh的toyopearlsp-650s和sp-650m,来自biorad的macro-prephighs,来自palltechnologies的ceramichyperds、trisacrylm和lssp以及spherodexlssp);基于磺乙基的基团(例如,来自emd的fractogelse,来自thermoscientific的poross-10和s-20);基于磺丙基的基团(例如,来自tosoh的tskgelsp5pw和sp-5pw-hr,来自thermoscientific的poroshs-20和hs50);基于磺基异丁基的基团(例如,来自emd的fractogelemdso3-);基于硫氧基乙基的基团(例如,来自whatman的se52、se53和express-ions);基于羧甲基的基团(例如,来自gehealthcare的cmsepharosefastflow,来自biochromlabsinc.的hydrocellcm,来自biorad的macro-prepcm,来自palltechnologies的ceramichyperdcm、trisacrylmcm、trisacryllscm,来自emd-millipore的matrxcellufinec500和c200,来自whatman的cm52、cm32、cm23和express–ionc,来自tosoh的toyopearlcm-650s、cm-650m和cm-650c);基于磺酸和羧酸的基团(例如来自j.t.baker的bakerbondcarboxy-sulfon);基于羧酸的基团(例如,来自j.tbaker的wpcbx,来自dowliquidseparations的dowexmac-3,来自sigma-aldrich的amberliteweakcationexchangers、dowexweakcationexchanger和diaionweakcationexchangers,和来自emd的fractogelemdcoo-);基于磺酸的基团(例如,来自biochromlabsinc.的hydrocellsp,来自dowliquidseparations的dowexfinemeshstrongacidcationresin,来自j.t.baker的unospheres、wpsulfonic,来自sartorius的sartobinds膜,来自sigma-aldrich的amberlitestrongcationexchangers、dowexstrongcation和diaionstrongcationexchanger);和基于正磷酸盐的基团(例如来自whatman的p11)。
含有感兴趣的蛋白质的混合物
本发明的方法可用于从含有一种或多种感兴趣的蛋白质的任何混合物中纯化所述蛋白质。在一个实施方案中,混合物从表达待纯化蛋白质的活细胞获得或由其产生(例如天然地或通过基因工程)。基因工程改造细胞以产生蛋白质的方法是本领域熟知的。参见,例如,ausabel等人编辑,(1990),currentprotocolsinmolecularbiology(wiley,newyork)以及美国专利no.5,534,615和4,816,567,将每篇文献通过提述明确并入本文。这样的方法包括引入编码进入活宿主细胞中的蛋白质的核酸并允许所述蛋白质的表达。这些宿主细胞可以是细菌细胞、真菌细胞,或优选培养物中生长的动物细胞。细菌宿主细胞包括但不限于大肠杆菌细胞。适合的大肠杆菌菌株的实例包括:hb101、dh5α、gm2929、jm109、kw251、nm538、nm539,和任何不能切割外来dna的大肠杆菌菌株。可以使用的真菌宿主细胞包括但不限于酿酒酵母、巴斯德毕赤酵母和曲霉菌属细胞。可以使用的动物细胞系的一些实例是cho、vero、dxb11、bhk、hela、cos、mdck、293、3t3、ns0和wi138。可以使用本领域技术人员熟知的方法(例如,通过转化、病毒感染和/或选择)建立新的动物细胞系。在其他实施方案中,感兴趣的蛋白质(例如抗体)在cho细胞中产生(参见,例如,wo94/11026)。本领域已知各种类型的cho细胞,例如cho-k1、cho-dg44、cho-dxb11、cho/dhfr-和cho-s。
混合物的制备最初取决于蛋白质的表达方式。一些细胞系统将蛋白质(例如抗体)从细胞直接分泌到周围的生长培养基中,而其他系统将抗体保留在细胞内。对于细胞内产生的蛋白质,可以使用多种方法中的任何一种破坏细胞,比如机械剪切、渗透压冲击和酶处理。所述破坏使细胞的全部内容物释放到匀浆中,并且另外产生可以通过离心或过滤除去的亚细胞片段。由于在蛋白质生产运行过程中细胞的自然死亡和细胞内宿主细胞蛋白的释放,产生了类似的关于直接分泌的蛋白质的问题,不过程度较小。
在一个实施方案中,例如从混合物中除去细胞或细胞碎片来制备澄清原液。本发明方法可采用任何适合的方法学来除去细胞或细胞碎片,包括离心、切向流过滤或深层过滤。
蛋白质纯化
本发明方法提供了用于从混合物中通过cex纯化感兴趣的蛋白质(例如抗体或fc融合蛋白)的改进技术。这些方法通常包括以下步骤:(a)提供包含感兴趣的蛋白质和一种或多种污染物的混合物;(b)将混合物上样到偶联有精氨酸的cex树脂上;和(c)从树脂中洗脱感兴趣的蛋白质,从而在cex色谱中以降低的聚集形成水平纯化感兴趣的蛋白质。在具体实施方案中,偶联有精氨酸的cex树脂是精氨酸-磺丙基(arg-sp)琼脂糖树脂。然而,技术人员将理解,可以在前述方法的步骤之前、之后或之间进行另外的纯化。与使用未偶联精氨酸的cex树脂相比,使用偶联有精氨酸的cex树脂导致感兴趣的蛋白质的聚集形成减少。
感兴趣的蛋白质(例如抗体)与阳离子交换树脂的结合可以在低于待纯化的所述蛋白质的最强酸性的同工型的pi的任何ph下进行。在具体的实施方案中,感兴趣的蛋白质在约ph4和约ph8之间(例如,约ph4、4.5、5、5.5、6、6.5、7、7.5和8)与树脂结合。在一个示例性实施方案中,感兴趣的蛋白质在约ph6.2与树脂结合。在另一个示例性实施方案中,感兴趣的蛋白质在约ph4.5与树脂结合。
一旦感兴趣的蛋白质与阳离子交换树脂结合,通过用缓冲液洗涤树脂除去污染物(例如hcp)。通过监测纯化蛋白质的纯度和产率,可以针对每种感兴趣的蛋白质凭经验确定用于洗涤树脂的最佳ph。可以用洗涤剂或表面活性剂(例如聚山梨醇酯)增强洗涤缓冲液,以进一步除去污染物,例如dna和内毒素污染物。
在洗涤阳离子交换树脂之后,使用缓冲液洗脱感兴趣的蛋白质。通常,通过增加洗脱缓冲液的ph或通过相对于结合缓冲液增加洗脱缓冲液的离子强度来促进洗脱,例如借助于向洗脱缓冲液中添加盐(例如氯化钠)。另外,可以将聚醚(例如,聚乙二醇)添加到洗脱缓冲液中以减少蛋白质聚集和更高分子量的物质的形成。
偶联有精氨酸的cex树脂的制备
在某些实施方案中,本发明提供了偶联有精氨酸的cex树脂。可以通过胺反应性化学物质以各种方式进行cex树脂与精氨酸的偶联,所述化学物质例如但不限于nhs酯、亚氨酸酯、五氟苯酯、羟甲基膦等。
如下面的工作实施例展示的,可以通过将3-氨基-1-丙磺酸的伯胺基与nhs活化的琼脂糖珠偶联来制备sp琼脂糖树脂。可以通过将l-精氨酸和3-氨基-1-丙磺酸依次偶联到nhs活化的琼脂糖珠上来制备arg-sp琼脂糖树脂。首先用di水彻底洗涤nhs活化的琼脂糖浆液以除去丙酮储存溶液,随后使用具有0.2μm滤纸的滤瓶,用0.1m磷酸钠、0.15mnacl的偶联缓冲液(ph7.2)洗涤。为了制备sp琼脂糖树脂,然后可以将琼脂糖珠与300mm3-氨基-1-丙磺酸以1:2的体积比在室温下混合2小时。为了制备arg-sp琼脂糖树脂,首先将琼脂糖珠与300mml-精氨酸混合5分钟,用偶联缓冲液洗涤,然后在室温下与300mm3-氨基-1-丙磺酸另外混合2小时。大约80%的偶联反应在最初0.5小时内发生。用偶联缓冲液充分洗涤配体固定的琼脂糖珠,然后加入1m乙醇胺(ph7.4)溶液将未反应的nhs官能团封端。最后,用偶联缓冲液再次洗涤功能化琼脂糖珠,用于柱填充。
通过下列实施例进一步展示了本公开,这些实施例不应该解释为进一步的限制。将所有附图和所有参考文献的内容、在本申请各处引证的专利和公开的专利申请都清楚地通过提述完整并入本文。
实施例1
关于了解在结构不稳定的fc融合蛋白的阳离子交换色谱中的聚集物形成和解离的见解
材料和方法
1.化学品、树脂和蛋白质
除非另有说明,否则所有化学品均来自j.t.baker(phillipsburg,nj,usa)。3-氨基-1-丙磺酸(3aps)和乙醇胺购自sigma-aldrich(st.louis,mo,usa)。spsepharosefastflow(spsff)和cmsepharosefastflow(cmsff)树脂获自gehealthcaresciences(uppsala,sweden)。porosxs树脂和n羟基琥珀酰亚胺(nhs)活化的琼脂糖浆液购自lifetechnologies(waltham,ma,usa)。unosphererapids和凝胶过滤标准品购自bio-rad(philadelphia,pa)。在这项工作中使用的fc融合蛋白在中国仓鼠卵巢(cho)细胞中表达,并且在bristol-myerssquibb,co.生产。这种fc融合蛋白的总分子量为78kda。通过ζ电位测量确定实验pi(等电点)为7.2。如前所述,对于这种fc融合蛋白,在铰链区中和在两条单链之间的其他位置没有二硫键。通过将蛋白a纯化的池缓冲交换到目标溶液条件下,获得在本研究中使用的蛋白质装载材料。
2.色谱仪器和方法
使用安装有unicorn软件版本6.3(piscataway,ny,usa)的gehealthcare
3.聚集物分析的sec方法
使用安装在waterscorporation(milford,ma,usa)的watershplc系统上的tosohbioscience(kingofprussia,pa,usa)的tskgelg3000swxl柱进行分析性sec。所述方法使用100mm磷酸钠、100mm硫酸钠(ph6.8),流速为1ml/min,恒定的总注入蛋白质量为100μg。通过uv280nm监测洗脱的蛋白质。
4.syproorange染料实验
将spsff、cmsff和porosxs树脂缓冲交换到25mm乙酸钠(ph5.0)缓冲液中并调节为50%(v/v)的重力沉降的浆液。对于含染料的样品,将400μl树脂浆液与98.5μl的25mm乙酸钠(ph5.0)缓冲液和1.5μl的购自invitrogen(paisley,scotland,u.k.)的syproorange染料(5000x)混合。对于非标记对照样品,将400μl树脂浆液与100μl25mm乙酸钠(ph5.0)缓冲液混合。将所有样品在辊式混合器上以180rpm倒转5分钟,然后在拍照之前使其经历重力沉降。
5.差示扫描荧光测定法(dsf)研究
将树脂spsff和cmsff缓冲交换到相应的缓冲液(50mm乙酸钠,ph4.0-5.5)中并调节为50%(v/v)的重力沉降的浆液。使用appliedbiosystems(warrington,cheshire,u.k.)的7500实时pcr(rt-pcr)系统(软件版本1.4.1)进行实验。简言之,将树脂(最终10%v/v)、蛋白质(最终1g/l)、syproorange染料(最终15x)和相应ph的缓冲液以预先计算的比例混合,并加入到快速光学96孔反应板(appliedbiosystems)中,至终体积20μl/孔,其中每个条件以一式两份制备。用购自appliedbiosystems的光学粘性膜密封后,在rt-pcr中直接分析所述板。加热循环包括持续2分钟的4℃预冷却步骤和随后从4℃至53℃梯度的99个步骤,其中每个步骤的升温率为每30秒0.5℃。使用检测syproorange染料的校准设置(λex490nm;λem580nm)收集数据,并通过将荧光数据拟合至修改的clarke和fersht方程进行分析。[35]
其中i(t)是荧光强度;t是每个荧光数据点的实际温度;tm是熔化温度;αf和βf分别是折叠状态的基线的截距和斜率;αa和βa分别是高温下荧光猝灭步骤的截距和斜率;m是与表观熔化温度下的转变的斜率相关的指数因子。通过使用最小二乘法来拟合dsf数据,获得αf、βf、αa、βa、m和tm的值。
6.内部树脂制备
通过将3-氨基-1-丙磺酸的伯胺基与nhs活化的琼脂糖珠偶联来制备sp琼脂糖树脂。通过将l-精氨酸和3-氨基-1-丙磺酸依次偶联到nhs活化的琼脂糖珠上来制备arg-sp琼脂糖树脂。首先用di水彻底洗涤nhs活化的琼脂糖浆液以除去丙酮储存溶液,随后使用具有0.2μm滤纸的滤瓶,用0.1m磷酸钠、0.15mnacl的偶联缓冲液(ph7.2)洗涤。为了制备sp琼脂糖树脂,然后将琼脂糖珠与300mm3-氨基-1-丙磺酸以1:2的体积比在室温下混合2小时。为了制备arg-sp琼脂糖树脂,首先将琼脂糖珠与300mml-精氨酸混合5分钟,用偶联缓冲液洗涤,然后在室温下与300mm3-氨基-1-丙磺酸另外混合2小时。根据供应商提供的方案,大约80%的偶联反应在最初0.5小时内发生。用偶联缓冲液充分洗涤配体固定的琼脂糖珠,然后加入1m乙醇胺(ph7.4)溶液将未反应的nhs官能团封端。最后,用偶联缓冲液再次洗涤功能化琼脂糖珠,用于柱填充。将这项工作中制备的arg-sp琼脂糖树脂填充到5mm内径的apminiglass柱(waterscorporation,milford,ma,usa)中,床高大约为5cm。
7.建模方法学
使用bioviadiscoverystudio软件中的同源建模获得蛋白质3d结构[36]。基于蛋白质结构,使用discoverystudio中的同源建模的结构,利用空间聚集倾向(sap)模型来确定易聚集的疏水区。蛋白质中每个原子的sap值定义如下[37],
在此,计算
基于discoverystudio软件中的蛋白质电离方案,计算不同ph条件下的蛋白质电荷。所述电荷基于预测蛋白质结构中每个可滴定氨基酸残基的pk1/2和滴定曲线。使用discoverystudio中的delphi程序计算静电势。delphi程序使用有限差分技术在立方晶格上求解poisson-boltzmann方程。在这些电荷和电势计算中使用charmm全原子力场[39]。通过利用计算的电势将分子表面着色将静电势可视化。通过对其组成原子的静电势值求平均来计算平均残基电势。
结果和讨论
1.fc融合蛋白在cex中的聚集行为
在使用50mm乙酸钠、250mmnacl(ph6.3)的盐步骤洗脱过程中,在spsff柱上观察到多峰洗脱图(图1a)。基于sec分析的洗脱物聚集物水平(8.6%)远高于装载材料(1.5%),并且指示了在cex步骤过程中显著的聚集物形成。图1a中的示踪剂显示在盐转变过程中的暂时ph下降(至<ph4.5),其与洗脱锋(front)同时出现,导致暂时的低ph和高盐环境。50mm乙酸钠、250mmnacl的洗脱缓冲液(ph5.6)显示出与ph6.3的乙酸盐相似的ph转变现象。在盐步骤洗脱中使用ph6.3的乙酸盐,以使蛋白质回收最大化,从而更好地定量所形成的聚集物。在其他研究中已经报道了在盐步骤洗脱过程中的类似的ph转变现象[32-34]。基于溶液内蛋白质稳定性研究,发现了低ph和高盐引起聚集和沉淀。一项单独的洗脱分级研究(未显示)也证实,晚期洗脱峰含有较高的聚集物水平,与之前的报道相似[12]。相反,使用ph8.3的50mm乙酸钠进行ph步骤洗脱,导致单峰洗脱和平稳的ph转变(图1b)。然而,在洗脱物中也观察到较多的聚集物形成(6.1%)。单峰洗脱图指示在ph步骤洗脱过程中形成的单体和聚集物具有相似的洗脱特性。基于在一项单独研究中评估的蛋白质溶液内稳定性(在室温下24小时)(数据未显示),蛋白质在低盐浓度下在ph4.0-8.0之间的溶液中是稳定的,并且在检查的条件下未观察到聚集物形成。这项研究提供了足够的证据,以排除蛋白质溶液性质是在ph步骤洗脱研究中观察到的聚集现象的根本原因。
为了更好地了解在两种不同的洗脱条件下形成的聚集物种类,将盐步骤和ph步骤洗脱池的sec曲线与装载材料(在ph4.5的50mm乙酸钠中)和使用50mm乙酸钠、250mmnacl在ph4.5制备的蛋白质溶液的sec曲线进行比较。选择蛋白质溶液样品的缓冲液条件以模拟洗脱时柱内缓冲液转变区中的缓冲液条件(图1a)。考虑到最大峰值处的蛋白质浓度和溶液条件随着柱中洗脱的进行而变化,并且缓冲液转变区中的蛋白质质量仅是总洗脱蛋白质的一部分,实际上不可能确定溶液样品与洗脱样品真正相当的条件。因此,蛋白质溶液样品只能提供在实际cex洗脱期间经历的条件的半定量比较。在sec图中观察到两个不同的峰,如图2所示。将这两个峰的保留时间与bio-rad的凝胶过滤标准品的色谱图进行比较表明,hmw1和hmw2分别大致对应于二聚体和寡聚物种类。寡聚物种类在溶液样品中是突出的,其在sec分析之前保持相当于cex步骤(图2)持续时间的时段(≤4小时)。在盐步洗脱物中,也观察到类似的sec图,表明洗脱时暂时的低ph和高盐环境也可引起蛋白质聚集并产生类似的hmw种类。盐步骤洗脱物中较低的聚集物水平可能是由于缓冲液转变区中洗脱物的盐浓度略低,如图1a所示。在ph步骤洗脱中形成的大多数聚集物是二聚体,其不同于在盐步骤洗脱过程中在低ph和高盐条件下形成的聚集物。由于ph洗脱不会对蛋白质产生不利的溶液环境,因此增加的聚集物水平很可能是cex中蛋白质-树脂相互作用的结果。
ph步骤洗脱用于评估蛋白质-树脂相互作用对在cex步骤过程中形成的聚集物性质的影响,因为这种洗脱模式可以有效避免蛋白质不稳定的溶液条件(例如,低ph和高盐)。使用这种方法,通过对保持在4℃的样品的定期sec分析来研究cex洗脱物中聚集物的稳定性。如图9所示,聚集物峰的大小随着保持时间的增加而降低,表明在spsff的cex步骤过程中形成的一些聚集物种类的可逆性质。在洗脱后的前8小时,洗脱物聚集物水平从6.1%降至4.1%,并且在5天后继续降低至2.1%。结果表明,聚集物解离动力学可以影响洗脱物聚集物含量的水平,所述含量实际上取决于样品保持时间。因此,cex中聚集物形成和聚集可逆性的促成因素的机械理解对于稳健且高产率的cex纯化方法的开发尤其重要。
2.提出的假设
不同于其他报道的研究[16-18],聚集物形成不需要结合蛋白的过度保持时间,并且洗脱物聚集物水平可能与时间依赖性自解离过程相关联。这里观察到的现象表明了一种复杂的聚集机制,从而产生这样的假设,即,在cex过程中形成的聚集物可能是由于具有可逆构象变化的那些分子之间暂时有利的蛋白质-蛋白质相互作用所致。值得注意的是,聚集物解离似乎是动力学驱动的过程,在此过程中天然构象在长达几天的时间尺度内逐渐恢复。
所提出的假设的示意图在图3中示出。所述假设描述了在cex吸附和解吸过程中的简单的两步聚集过程以及在cex洗脱物中的聚集物解离过程。根据所提出的聚集机制,在cex洗脱物中的聚集物水平(air+ar)是两个促成因素的结果:结合蛋白的构象变化和解吸后这些部分解折叠分子
3.机械研究
1)柱装载的影响
研究了柱装载对spsff洗脱物中聚集物水平的影响。洗脱后立即测定所有洗脱物样品的sec,并在5天后再次测定(在4℃下储存)。t=0天时的洗脱物聚集物水平在相对宽范围的柱装载(1-50g/l树脂)内显著变化。spsff的动态结合能力大于50g/l树脂。在这里将不同装载条件下的sec结果直接比较,因为所有实验都具有很高的总产品回收率(≥98%)。如图4所示,洗脱物聚集物水平首先增加,然后略微降低,其中最大聚集物水平达到大约30g/l树脂。最终,所有洗脱物样品在5天(t=5天)后显示实际上相同的聚集物水平。结果表明柱装载对初始聚集物水平的影响,而spsff洗脱物中不可逆聚集物的量似乎基本上与测试的特定条件下的装载条件无关。通过将吸附过程中的蛋白质构象变化
2)装载ph的影响
在spsff上进行一系列实验,以评估装载ph(4.0至5.5)对洗脱物聚集物水平的影响。不同ph条件下的装载电导率仅在很小的范围内(2.0-2.5ms/cm)变化。表1显示,洗脱物聚集物水平随着装载ph的增加而显著降低,这是与总的吸引的蛋白质-树脂相互作用一致的趋势。使用在方法部分中描述的同源建模工具计算蛋白质的静电势,以说明在不同ph条件下的电荷分布。电荷建模(图10)指示,尤其是在cdr和fc区中的蛋白质表面随着ph增加而表现出更多的带负电荷的片区。当与树脂上的阴离子配体相互作用时,蛋白质上的这些负电荷表面区域可以产生排斥环境,从而降低总结合强度和蛋白质构象变化。此外,图11中的电荷电势变化清楚地显示了在小的ph变化后氨基酸残基的电荷电势经历剧烈变化的位置。chang和lenhoff报道[43],蛋白质-树脂相互作用可以在很大程度上取决于蛋白质结构中的少量氨基酸残基,这可能有助于解释装载ph条件对保持结合蛋白的构象完整性并因此降低洗脱物聚集物水平的影响。chaudhri等人[44]使用粗粒度计算模型并且证实mab-mab结合倾向在仅少数氨基酸不同的突变体中可以显著不同。鉴定对cex中的蛋白质结构稳定性特别重要的特定氨基酸残基可有助于通过在蛋白质一级序列中进行生物学上不重要的改变来工程改造抗聚集的分子[45][46]。显然,制造友好的生物分子的合理设计需要另外的建模工作来解释蛋白质-蛋白质和蛋白质-树脂相互作用对潜在蛋白质构象变化的影响,这超出了这项工作的范围。
表1.装载ph对洗脱物聚集物水平的影响(ph步骤洗脱)。在不同ph条件下以30g/l树脂装载spsff柱。洗脱后立即进行sec分析。
作为正交实验工具,dsf用于进一步研究蛋白质构象变化的存在和严重性,并将cex条件与洗脱物聚集物水平相关联。使用syproorange染料进行dsf研究,所述染料是一种部花菁(merocyamine)染料的成员,当与研究对象(例如蛋白质)的疏水表面结合时,其可以激活荧光[47-49]。在不同ph下与cm和spsepharose结合的蛋白质的dsf结果显示在图12中。将记录随着逐渐升高温度的制备样品的荧光的稳定性曲线拟合至方程式1,以估算熔化温度(tm)。由于tm描述了蛋白质构象完整性的热稳定性,因此在此使用它作为在检测的溶液条件下的蛋白质构象稳定性的替代指示物。
如表2所示,结合蛋白和游离蛋白两者的拟合tm值都随ph降低而降低,表明蛋白质在较低ph下变得构象较不稳定。在游离溶液中,蛋白质的tm值仅显示从ph5.5至ph4.5的非常小的降低,但是在ph4时其显著降低,其中蛋白质可能具有降低的结构刚性和/或经历较大的构象变化。对于结合蛋白,两种树脂的tm的显著降低发生在ph4.5-5.0附近的较高ph下。结果清楚地表明,在相应的条件下,结合蛋白暴露更多的疏水区域并且变得比在游离溶液中在构象上更不稳定。此外,结合蛋白在spsff上的tm值似乎始终低于其在cmsff上的tm值,表明当与spsff结合时蛋白质的结构稳定性较低。这个观察到的模式可能与总体配体性质(例如,类型、密度、接头柔性等)相关,因为两种树脂具有相同的琼脂糖基础基质和相似的孔形态[50]。稍后将更详细地论述不同树脂的cex行为。将电荷建模与dsf结果相结合,可以看出在较低装载ph下增加的结合强度导致更高程度的蛋白质构象变化、更疏水的表面暴露和更低的蛋白质结构稳定性,这进一步导致cex中更多的聚集物形成。如图13所示,装载ph与计算和测量的生物物理特性以及cex洗脱物聚集物水平相当好地相关。
表2.在不同ph条件在cex树脂存在和不存在的情况下根据dsf结果拟合的熔化温度(tm)
aspsff和cmsff的树脂装载量为10g/l树脂
对于所研究的蛋白质,cex步骤中表现出的构象不稳定性可归因于两条单链之间二硫键的缺乏。据报道,虽然蛋白质二级和三级结构在很大程度上仍然保留在cex树脂表面上,也可发生局部构象变化[16]。这里采用sap建模来探索小的构象变化对蛋白质疏水性的潜在影响。这个模型估算溶剂暴露的蛋白质表面上的疏水区域的范围[37]。假定简单的情况是,在蛋白质吸附到树脂表面上后,两条单链可以彼此分开几个埃
3)装载过程中停留时间(rt)的影响
研究了装载rt对洗脱物聚集物水平的影响,以探索结合蛋白中构象变化发生的时间尺度。通过改变装载rt(即流速)和/或在将蛋白质装载到cex柱上之后增加静态保持来改变总蛋白质/树脂接触时间。对于本研究,洗脱rt保持恒定。如表3所示,当装载rt从2分钟变为6分钟时,洗脱物聚集物水平从4.5%适度地增加至6.1%,这对应于结合蛋白/树脂接触时间从12分钟增加至36分钟。更长的装载rt不仅可以由于更长的接触时间而导致更多的蛋白质构象变化,而且由于在cex过程中颗粒内质量转移的作用不太显著而导致洗脱后更高的蛋白质浓度,这两者都能促成洗脱物聚集物水平的增加。这种现象与将蛋白质-蛋白质相互作用、蛋白质构象稳定性和聚集率关联为蛋白质浓度的函数的溶液研究很好地一致[26]。值得注意的是,随着6分钟的装载rt和另外12小时的静态保持,洗脱物聚集物水平增加至17.7%,表明蛋白质/树脂接触时间对引起fc融合蛋白的剧烈构象变化的显著影响。在别处也报道了针对igg1和igg2的这种观察结果[12,18]。应该注意的是,吸附后经历结构变化的蛋白质的动力学时间尺度实际上与正常的cex操作相关。因此,装载rt是帮助控制cex步骤中洗脱物聚集物水平的关键因素,应尽可能避免对结构不稳定的蛋白质进行缓慢装载和不必要的保持,就像本研究中所研究的那样。还强调了在cex工艺开发过程中测试广泛范围的蛋白质/树脂接触时间的重要性,从而即使具有意外的结合产物的柱上保持,也能确保稳健的柱性能。有趣的是,在具有实际相关性的不同洗脱rt条件下,未观察到洗脱物聚集物水平的重大差异(数据未显示)。这表明洗脱过程中的聚集动力学比吸附后经历构象变化的蛋白质的动力学快得多。
表3.装载过程中停留时间对ph步骤洗脱中洗脱物聚集物水平的影响。在ph4.5以30g/l树脂装载spsff柱。洗脱后立即进行sec分析。
4)cex树脂类型的影响
鉴于蛋白质结构变化几乎总是不可避免地导致蛋白质表面疏水性的变化,在相似条件下研究了spsff、cmsff和porosxs以评估树脂特性的影响,特别是固定相疏水性和可电离配体类型。porosxs和spsff的支持基质分别为交联的聚(苯乙烯-二乙烯基苯)和交联的琼脂糖。cmsff具有与spsff相同的基础基质,但树脂用co3-配体功能化。
如图6a所示,初始洗脱物聚集物水平在三种树脂中明显不同,porosxs(7.2%)、spsff(6.1%)和cmsff(3.7%)按降序排列。应该注意的是,porosxs的蛋白质回收率仅为89%,远低于spsff(≥98%)和cmspp(≥98%)。装载的剩余蛋白质只能在苛性碱清洗步骤过程中从porosxs柱中除去,这表明非常强的蛋白质-树脂相互作用,其导致实际上不可逆的结合。为了更好地了解树脂特性的差异,在表4中列出的条件下将syproorange染料溶液与新鲜树脂颗粒混合。如图14所示,基于在添加染料之前和之后树脂相中的光色变化,cmsff和spsff显示非常少的染料结合,这可能主要是由于琼脂糖基础基质的已知亲水性质所致。相反,porosxs显示出显著的染料结合,如树脂颜色的变化所示。这清楚地表明,尽管其具有亲水涂层,但porosxs的疏水性仍然足以结合疏水性染料。与cmspp和spsff相比,porosxs可以通过蛋白质和树脂之间的二级疏水相互作用与结合蛋白更强地相互作用,这也解释了porosxs洗脱物中不可逆结合和聚集物形成的更高百分比。然而,应该注意的是,难以将观察到的趋势归因于特定因素,因为不同的配体密度、接头性质、孔结构和基础基质的疏水性的影响是不同树脂的混淆变量。
表4.syproorange染料实验的标记条件
图6中的另一观察结果是所研究的三种树脂的聚集可逆性的比较。如图6a所示,cmsff和spsff的洗脱物聚集物水平降低至小于2%,而对于porosxs即使在120小时之后仍然大约为4%。此外,如图6b所示,在porosxs洗脱过程中形成的聚集物以显著更小的程度(热力学方面)和慢得多的速率(动力学方面)恢复为单体。cmsff和spsff洗脱物中的聚集物显示出相似程度的可逆性,表明可能相似的生物物理特性。porosxs与其他两种树脂之间聚集可逆性的明显差异反映了porosxs上强得多的总蛋白质-树脂相互作用(包括蛋白质/配体和蛋白质/树脂表面),其导致形成的聚集物种类的不同分子特性。
4.提出的机制的实验验证
已经测试了各种策略,以减少或防止cex步骤中的聚集物形成。发现使用没有接枝聚合物的大孔树脂(比如unosphererapid)可有效减少cex诱导的产物解折叠和mab的聚集[17]。然而,这种树脂在这项工作中评价的条件下不能减轻聚集物形成(数据未显示)。向装载/洗涤/洗脱溶液中添加赋形剂(例如精氨酸、甘氨酸和蔗糖)也被证明是无效的。这些结果意味着与其他早期研究不同的聚集机制。根据提出的假设,聚集物形成是洗脱后结合蛋白的构象变化的严重性和易聚集蛋白的浓度的结果。因此,诸如高装载ph、短的蛋白质/树脂接触时间、亲水性树脂表面和弱可电离配体的措施可以帮助减少聚集物形成。然而,在这项研究中,这些努力都未能完全阻止聚集物形成。
除了上面评估的易于应用的措施之外,还进行了树脂修饰以检验所提出的聚集假设并且在分子水平上获得对聚集现象的机械理解。以相对低的sp配体密度在内部制备了定制树脂(用sp和arg-sp配体功能化),其中(有和没有)将精氨酸另外固定到树脂表面以产生接近树脂表面的二级蛋白质-精氨酸相互作用。在相同条件下测量时,完全功能化的sp琼脂糖树脂的动态结合能力为30g/l树脂,其低于spsff的动态结合能力(≥50g/l树脂)。由于固定化精氨酸的弱化作用,制备的arg-sp琼脂糖树脂的动态结合能力为20g/l树脂。尽管其广泛用作溶液中的蛋白质稳定剂,但尚未将精氨酸固定到树脂表面上来调节具有永久性二级蛋白质-精氨酸相互作用的主要蛋白质-树脂吸引力。图7显示了arg-sp修饰的混合模式琼脂糖珠的示意图。
包括两种定制树脂在内的几种不同树脂的比较如图8所示。可以看出,与spsff相比,sp琼脂糖树脂的聚集物形成较低。这被认为主要是由于定制的sp琼脂糖树脂中配体密度相对较低所致,正如最近的分子级研究表明,其中在不同聚类条件下的随机配体分布导致不同的蛋白质吸附特性,即使对于化学上相同的配体也是如此[51]。较低的配体密度导致每结合蛋白的相互作用配体的数目更少,并有助于在一定程度上减少聚集物形成。对于arg-sp琼脂糖树脂,在洗脱物中未观察到明显的聚集物增加。这可能是由于固定化精氨酸对蛋白质-树脂相互作用的弱化作用所致。固定化精氨酸实际上可以用作混合电荷配体(即带负电荷的羧基和带正电荷的胍基),其可以阻碍结合蛋白和树脂表面之间的不希望的相互作用。固定化精氨酸中的带正电荷的胍基团可促成更弱的结合强度和更少的蛋白质构象变化。值得注意的是,精氨酸对蛋白质-树脂相互作用的弱化作用仅在精氨酸被固定到树脂表面上时才有效,在树脂表面上它起着混合模式配体而不是游离溶质的作用。精氨酸影响的差异可以表明使用精氨酸作为混合模式配体来减轻cex中结构不稳定的蛋白质的聚集问题的独特优势。尚不清楚固定化精氨酸上的胍基团是否具有类似于向蛋白质溶液中添加精氨酸时所观察到的另外的蛋白质稳定作用[52]。这个问题的答案超出了这项工作的范围,并且需要另外的研究,其涉及用适当选择的官能团固定不同的分子。虽然固定程序正在进一步优化(单独的努力)以提高结合能力并保持hmw减轻能力,但在这里获得的结果为所提出的假设提供了支持性证据。
结论
这项工作研究了cex步骤中结构不稳定的fc融合物的聚集行为。对于使用盐步骤洗脱的多峰洗脱图,发现其与在柱内缓冲液转变过程中由蛋白质稳定性特性引起的聚集物形成相关。通过使用ph步骤洗脱实现单峰洗脱,而通过源自结合蛋白的构象变化的不同机制发生聚集。ph步骤洗脱数据明确显示了cex结合/洗脱过程对在分子稳定的溶液条件下的蛋白质构象稳定性和聚集倾向的影响。形成的聚集物可以是热力学上不利的,并且在所研究的条件下在几天之内部分地恢复为天然构象。相对较慢的聚集物解离动力学强调了正确且一致的采样、样品储存和分析测试实践的重要性,以确保有代表性且可再现的分析结果。发现聚集物形成对cex柱装载是敏感的,所述cex柱装载影响结合蛋白的构象变化的严重性和洗脱后易聚集蛋白的浓度。这两个因素共同促成了作为柱装载的函数的洗脱物聚集物水平的非线性模式。
cex洗脱物中的聚集物形成和稳定性也可受其他操作条件(例如ph、流速等)以及树脂特性的影响。通常,降低蛋白质结合强度和蛋白质-树脂接触时间的那些实际措施可导致由于结合蛋白的较少构象变化所致的较低的聚集物水平,如dsf结果所证实。优选的树脂特性包括弱的可电离配体(例如cm功能性)、亲水性树脂表面和较低的配体密度。使用内部制备的arg-sp琼脂糖树脂的研究为针对cex步骤中所见的聚集行为提出的假设提供了实验验证。将精氨酸固定到树脂表面上即使在低ph条件下也有效地使蛋白质和树脂之间的不希望的相互作用最小化,而在低ph条件的情况下,蛋白质表现出使用所有其他cex树脂评价的结构不稳定性和高洗脱物聚集物水平。这项工作不仅为增强对cex步骤中的结构不稳定的fc融合蛋白的聚集物形成的机械理解提供了线索,而且为特殊应用的新树脂设计提供了一些有趣的方向,比如在这里研究的应用。最后,在溶液中观察到的蛋白质稳定性特性与其在色谱过程中的聚集倾向之间的不一致性强调了获取分子的下游可加工性信息(即,本研究)在设计制造友好的生物分子和开发稳健的cex精制工艺中的重要性。
参考文献:
[1]j.m.reichert,monoclonalantibodiesasinnovativetherapeutics,curr.pharm.biotechnol.9(2008)423-430.
[2]d.m.ecker,s.d.jones,h.l.levine,thetherapeuticmonoclonalantibodymarket,mabs7(2015)9-14.
[3]z.chen,t.chen,x.sun,b.j.hinds,dynamicelectrochemicalmembranesforcontinuousaffinityproteinseparation,adv.funct.mater.24(2014)4317-4323.
[4]a.a.shukla,b.hubbard,t.tressel,s.guhan,d.low,downstreamprocessingofmonoclonalantibodies--applicationofplatformapproaches,j.chromatogr.b848(2007)28-39.
[5]a.staby,m.b.sand,r.g.hansen,j.h.jacobsen,l.a.andersen,m.gerstenberg,u.k.bruus,i.h.jensen,comparisonofchromatographicion-exchangeresinsiv.strongandweakcation-exchangeresinsandheparinresins,j.chromatogr.a1069(2005)65-77.
[6]l.yu,l.zhang,y.sun,proteinbehavioratsurfaces:orientation,conformationaltransitionsandtransport,j.chromatogr.a1382(2015)118-134.
[7]a.jungbauer,c.machold,r.hahn,hydrophobicinteractionchromatographyofproteins.iii.unfoldingofproteinsuponadsorption,j.chromatogr.a1079(2005)221-228.
[8]y.xiao,a.rathore,j.p.o'connell,e.j.fernandez,generalizingatwo-conformationmodelfordescribingsaltandtemperatureeffectsonproteinretentionandstabilityinhydrophobicinteractionchromatography,j.chromatogr.a1157(2007)197-206.
[9]l.zhang,g.zhao,y.sun,molecularinsightintoproteinconformationaltransitioninhydrophobicchargeinductionchromatography:amoleculardynamicssimulation,j.phys.chem.b113(2009)6873-6880.
[10]a.voitl,a.butte,m.morbidelli,behaviorofhumanserumalbuminonstrongcationexchangeresins:ii.modelanalysis,j.chromatogr.a1217(2010)5492-5500.
[11]a.voitl,a.butte,m.morbidelli,behaviorofhumanserumalbuminonstrongcationexchangeresins:i.experimentalanalysis,j.chromatogr.a1217(2010)5484-5491.
[12]r.gillespie,t.nguyen,s.macneil,l.jones,s.crampton,s.vunnum,cationexchangesurface-mediateddenaturationofanaglycosylatedimmunoglobulin(igg1),j.chromatogr.a1251(2012)101-110.
[13]t.arakawa,d.ejima,k.tsumoto,n.obeyama,y.tanaka,y.kita,s.n.timasheff,suppressionofproteininteractionsbyarginine:aproposedmechanismofthearginineeffects,biophys.chem.127(2007)1-8.
[14]u.das,g.hariprasad,a.s.ethayathulla,p.manral,t.k.das,s.pasha,a.mann,m.ganguli,a.k.verma,r.bhat,s.k.chandrayan,s.ahmed,s.sharma,p.kaur,t.p.singh,a.srinivasan,inhibitionofproteinaggregation:supramolecularassembliesofarginineholdthekey,plosone,2(2007)e1176.
[15]m.t.gao,x.y.dong,y.sun,interactionsbetweenl-arginine/l-argininederivativesandlysozymeandimplicationstotheirinhibitioneffectsonproteinaggregation,biotechnol.prog.29(2013)1316-1324.
[16]j.guo,g.carta,unfoldingandaggregationofaglycosylatedmonoclonalantibodyonacationexchangecolumn.partii.proteinstructureeffectsbyhydrogendeuteriumexchangemassspectrometry,j.chromatogr.a1356(2014)129-137.
[17]j.guo,g.carta,unfoldingandaggregationofmonoclonalantibodiesoncationexchangecolumns:effectsofresintype,loadbuffer,andproteinstability,j.chromatogr.a1388(2015)184-194.
[18]j.guo,s.zhang,g.carta,unfoldingandaggregationofaglycosylatedmonoclonalantibodyonacationexchangecolumn.parti.chromatographicelutionandbatchadsorptionbehavior,j.chromatogr.a1356(2014)117-128.
[19]h.luo,n.macapagal,k.newell,a.man,a.parupudi,y.li,y.li,effectsofsalt-inducedreversibleself-associationontheelutionbehaviorofamonoclonalantibodyincationexchangechromatography,j.chromatogr.a1362(2014)186-193.
[20]h.luo,m.cao,k.newell,c.afdahl,j.wang,w.k.wang,y.li,double-peakelutionprofileofamonoclonalantibodyincationexchangechromatographyiscausedbyhistidine-protonation-basedchargevariants,j.chromatogr.a1424(2015)92-101.
[21]w.wang,s.singh,d.l.zeng,k.king,s.nema,antibodystructure,instability,andformulation,j.pharm.sci.96(2007)1-26.
[22]a.l.fink,l.j.calciano,y.goto,t.kurotsu,d.r.palleros,classificationofaciddenaturationofproteins:intermediatesandunfoldedstates,biochemistry,33(1994)12504-12511.
[23]j.buchner,m.renner,h.lilie,h.j.hinz,r.jaenicke,t.kiefhabel,r.rudolph,alternativelyfoldedstatesofanimmunoglobulin,biochemistry,30(1991)6922-6929.
[24]s.b.hari,h.lau,v.i.razinkov,s.chen,r.f.latypov,acid-inducedaggregationofhumanmonoclonaligg1andigg2:molecularmechanismandtheeffectofsolutioncomposition,biochemistry,49(2010)9328-9338.
[25]r.f.latypov,s.hogan,h.lau,h.gadgil,d.liu,elucidationofacid-inducedunfoldingandaggregationofhumanimmunoglobulinigg1andigg2fc,j.biol.chem.287(2012)1381-1396.
[26]r.ghosh,c.calero-rubio,a.saluja,c.j.roberts,relatingprotein-proteininteractionsandaggregationratesfromlowtohighconcentrations,j.pharm.sci.105(2016)1086-1096.
[27]c.j.roberts,non-nativeproteinaggregationkinetics,biotechnol.bioeng.98(2007)927-938.
[28]e.j.yearley,p.d.godfrin,t.perevozchikova,h.zhang,p.falus,l.porcar,m.nagao,j.e.curtis,p.gawande,r.taing,i.e.zarraga,n.j.wagner,y.liu,observationofsmallclusterformationinconcentratedmonoclonalantibodysolutionsanditsimplicationstosolutionviscosity,biophys.j.106(2014)1763-1770.
[29]s.yadav,t.m.laue,d.s.kalonia,s.n.singh,s.j.shire,theinfluenceofchargedistributiononself-associationandviscositybehaviorofmonoclonalantibodysolutions,mol.pharm.9(2012)791-802.
[30]a.chaudhri,i.e.zarraga,t.j.kamerzell,j.p.brandt,t.w.patapoff,s.j.shire,g.a.voth,coarse-grainedmodelingoftheself-associationoftherapeuticmonoclonalantibodies,j.phys.chem.b116(2012)8045-8057.
[31]j.l.fast,a.a.cordes,j.f.carpenter,t.w.randolph,physicalinstabilityofatherapeuticfcfusionprotein:domaincontributionstoconformationalandcolloidalstability,biochemistry,48(2009)11724-11736.
[32]s.ghose,t.m.mcnerney,b.hubbard,phtransitionsinion-exchangesystems:roleinthedevelopmentofacation-exchangeprocessforarecombinantprotein,biotechnol.prog.18(2002)530-537.
[33]t.m.pabst,g.carta,phtransitionsincationexchangechromatographiccolumnscontainingweakacidgroups,j.chromatogr.a1142(2007)19-31.
[34]j.s.perez,d.d.frey,behavioroftheinadvertentphtransientformedbyasaltgradientintheion-exchangechromatographyofproteins,biotechnol.prog.21(2005)902-910.
[35]f.wang,s.sen,y.zhang,i.ahmad,x.zhu,i.a.wilson,v.v.smider,t.j.magliery,p.g.schultz,somatichypermutationmaintainsantibodythermodynamicstabilityduringaffinitymaturation,proc.natl.acad.sci.usa110(2013)4261-4266.
[36]bioviasoftwareinc.,discoverystudiomodelingenvironment,release4.1,sandiego:bioviasoftwareinc.,2014.
[37]n.chennamsetty,v.voynov,v.kayser,b.helk,b.l.trout,designoftherapeuticproteinswithenhancedstability,proc.natl.acad.sci.usa106(2009)11937-11942.
[38]s.d.black,d.r.mould,developmentofhydrophobicityparameterstoanalyzeproteinswhichbearpost-orcotranslationalmodifications,anal.biochem.193(1991)72-82.
[39]a.d.mackerell,d.bashford,m.bellott,r.l.dunbrack,j.d.evanseck,m.j.field,s.fischer,j.gao,h.guo,s.ha,d.joseph-mccarthy,l.kuchnir,k.kuczera,f.t.lau,c.mattos,s.michnick,t.ngo,d.t.nguyen,b.prodhom,w.e.reiher,b.roux,m.schlenkrich,j.c.smith,r.stote,j.straub,m.watanabe,j.wiorkiewicz-kuczera,d.yin,m.karplus,all-atomempiricalpotentialformolecularmodelinganddynamicsstudiesofproteins,j.phys.chem.b102(1998)3586-3616.
[40]d.s.tomar,s.kumar,s.k.singh,s.goswami,l.li,molecularbasisofhighviscosityinconcentratedantibodysolutions:strategiesforhighconcentrationdrugproductdevelopment,mabs,8(2016)216-228.
[41]r.chaudhuri,y.cheng,c.r.middaugh,d.b.volkin,high-throughputbiophysicalanalysisofproteintherapeuticstoexamineinterrelationshipsbetweenaggregateformationandconformationalstability,aapsj.16(2014)48-64.
[42]e.y.chi,s.krishnan,t.w.randolph,j.f.carpenter,physicalstabilityofproteinsinaqueoussolution:mechanismanddrivingforcesinnonnativeproteinaggregation,pharm.res.20(2003)1325-1336.
[43]c.chang,a.m.lenhoff,comparisonofproteinadsorptionisothermsanduptakeratesinpreparativecation-exchangematerials,j.chromatogr.a827(1998)281-293.
[44]a.chaudhri,i.e.zarraga,s.yadav,t.w.patapoff,s.j.shire,g.a.voth,theroleofaminoacidsequenceintheself-associationoftherapeuticmonoclonalantibodies:insightsfromcoarse-grainedmodeling,j.phys.chem.b117(2013)1269-1279.
[45]j.m.perchiacca,c.c.lee,p.m.tessier,optimalchargedmutationsinthecomplementarity-determiningregionsthatpreventdomainantibodyaggregationaredependentontheantibodyscaffold,proteineng.des.sel.27(2014)29-39.
[46]s.j.shire,formulationandmanufacturabilityofbiologics,curr.opin.biotechnol.20(2009)708-714.
[47]f.h.niesen,h.berglund,m.vedadi,theuseofdifferentialscanningfluorimetrytodetectligandinteractionsthatpromoteproteinstability,nat.protoc.2(2007)2212-2221.
[48]c.r.c.reichardt,solventsandsolventeffectsinorganicchemistry,vch,weinheim,1988.
[49]f.vollrath,n.hawkins,d.porter,c.holland,m.boulet-audet,differentialscanningfluorimetryprovideshighthroughputdataonsilkproteintransitions,sci.rep.4(2014)5625.
[50]p.dephillips,a.m.lenhoff,poresizedistributionsofcation-exchangeadsorbentsdeterminedbyinversesize-exclusionchromatography,j.chromatogr.a883(2000)39-54.
[51]l.kisley,j.chen,a.p.mansur,b.shuang,k.kourentzi,m.-v.poongavanam,w.-h.chen,s.dhamane,r.c.willson,c.f.landes,unifiedsuperresolutionexperimentsandstochastictheoryprovidemechanisticinsightintoproteinion-exchangeadsorptiveseparations,proc.natl.acad.sci.usa111(2014)2075-2080.
[52]b.m.baynes,d.i.wang,b.l.trout,roleofarginineinthestabilizationofproteinsagainstaggregation,biochemistry,44(2005)4919-4925.
- 还没有人留言评论。精彩留言会获得点赞!