雌甾-4,9-二烯-3,17-二酮的制备方法与流程
本发明涉及一种米菲司酮中间体的制备方法,具体涉及一种雌甾-4,9-二烯-3,17-二酮的制备方法。
背景技术:
米非司酮是一种新型的子宫内膜孕酮拮抗剂,具有终止早孕、抗着床、抑制排卵及促进子宫成熟的作用,是一种很好的口服避孕、抗早孕以及治疗子宫内膜异位症的药物。而雌甾-4,9-二烯-3,17-二酮是合成米菲司酮的重要中间体。
目前制备米非司酮中间体雌甾-4,9-二烯-3,17-二酮的方法主要包括以下几种:第一种方法、以薯芋皂苷为原料,经200℃加压裂解、氧化、水解得到醋酸双烯妊娠孕酮,再经beckmann重排、加成反应、取代反应、消除反应、水解反应、氧化反应、水解反应、氧化反应、脱羧反应、加成反应和消除反应等十多步反应制得最终产物4,9-雌甾二烯-3,17-二酮。此法以本身含有四个甾体母环的皂素为基础原料,成本高,环境污染大,且在进行肟化、贝克曼重排的反应中溶剂及水的用量较大,对温度比较敏感,不易控制,收率不高,此外反应需要的三氯氧磷属于受管制的剧毒化学品。第二种方法、以2,3,7,7α-四氢-1h-茚-1,5(6h)二酮(cn108003210a)为原料,经羰基还原反应、羟基保护反应、取代反应、双键还原反应、缩酮水解反应、关环反应、取代反应、缩酮水解反应、关环反应、羟基脱保护反应和羟基氧化反应十多步,反应路线较长且繁琐。
但是,目前制备米非司酮中间体雌甾-4,9-二烯-3,17-二酮的方法都存在合成过程中操作步骤繁琐,有机溶剂用量大,对环境污染大等问题。
技术实现要素:
为解决上述问题,本发明实施例提供了一种制备雌甾-4,9-二烯-3,17-二酮的新方法。
本发明实施例提供的雌甾-4,9-二烯-3,17-二酮的制备方法包括以下步骤:
以具有通式i的化合物为反应原料,对所述反应原料依次进行混合酸酐制备、格氏反应、闭环反应、水解及闭环反应,制得具有结构式v的化合物;
所述具有结构式v的化合物的合成路线如下:
优选的,所述混合酸酐制备的反应过程包括:
将使用第一有机溶剂溶解所述反应原料中,得到第一溶液,将所述第一溶液降温至第一预设温度并保温第一预设时长;
向所述第一溶液中缚酸剂和特戊酰氯,反应生成具有通式ii的化合物,反应结束后得到包含所述具有通式ii的化合物的第二溶液。
优选的,所述反应原料、所述缚酸剂和所述特戊酰氯之间的重量份数的比例为1:(0.72~1.62:):(1~1.1)。
优选的,所述第一有机溶剂包括四氢呋喃、无水乙醚和甲基四氢呋喃中的至少一种;
所述缚酸剂包括吡啶、三乙胺和二苯甲酮中的至少一种。
优选的,所述第一预设温度为-30℃~-40℃;
所述第一预设时长为2~4h。
优选的,所述格氏反应的反应过程包括:
在氮气保护下,将包含所述具有通式ii的化合物的第二溶液降温至第二预设温度,滴加格氏试剂,滴加操作完成后进行保温反应,所述保温反应的反应时长为第二预设时长,所述保温反应结束后对所得的反应体系进行终止反应和后处理,得到包含具有结构式iii的化合物的第三溶液。
优选的,所述第二预设温度为-20℃~-60℃;
所述第二预设时长为3~5h。
优选的,所述终止反应的反应过程包括:
向所述反应体系中加入反应终止液,所述反应终止液包括氯化铵水溶液、盐酸和醋酸水溶液中的至少一种。
优选的,所述闭环反应的反应过程包括:
使用第二有机溶剂溶解所述具有结构式iii的化合物,向所得的溶液中加入碱性固体和水的组合物或所述碱性固体,进行闭环反应,待所述闭环反应结束后得到具有结构式iv的化合物。
优选的,所述闭环反应中,所述第二有机溶剂包括四氢呋喃、甲醇、异丙醇和甲苯中的至少一种;
所述碱性固体包括氢氧化钾、叔丁醇钾、碳酸钠和氢氧化钠中的至少一种。
优选的,所述闭环反应步骤中,所述闭环反应的反应温度为所述第二有机溶剂的回流温度;
所述闭环反应的反应时间为10~12h。
优选的,所述闭环反应的反应过程还包括:
对包含所述具有结构式iv的化合物的反应液进行静置分层,分离得到有机相和水相;
对所述有机相和所述水相进行后处理。
所述后处理的步骤包括:
(1)将洗液加入所述有机相中,洗去所述有机相中的杂质,所述洗液包括氯化钠溶液、碳酸氢钠溶液和碳酸钠溶液中的一种或多种;
(2)将萃取剂加入上述步骤(1)所得的所述水相中,用来萃取所述水相中残留的有机相,所述萃取剂包括甲苯、四氢呋喃和甲醇中的一种或多种;
(3)对上述步骤(2)所得的萃取相进行浓缩处理,以去除所述萃取相中的萃取剂;对所述有机相进行浓缩,以除去有机相中的有机溶剂。浓缩结束,合并萃取相和有机相的浓缩液,并加入环己烷、丙酮、正己烷和甲醇中的一种或多种溶剂;
(4)对上述步骤(3)所得的已加入所述溶剂的萃取相和有机相浓缩液进行冰水浴降温,以析出固体,搅拌时间为2~6h;
(5)将上述步骤(4)所得的冰水浴降温后的混合物进行过滤,并对所得的固体进行干燥,得到所述具有结构式iv的化合物固体,所述干燥的干燥温度为50℃~70℃以及干燥时间为2~4h。
优选的,所述闭环反应中,所述具有结构式iv的化合物与所述碱固体之间的重量份数的比例为1:(0.47~0.77)。
优选的,所述水解及闭环反应的反应过程包括:
步骤a、将所述具有结构式iv的化合物、盐酸和第三有机溶剂混合,进行第一保温反应,待所述第一保温反应结束后向所得的第一反应液中加入第一终止液,得到水解产物中间体;
步骤b、将所述水解产物中间体、碱性试剂和第四有机溶剂混合,进行第二保温反应,待所述第二保温反应结束后向所得的第二反应液中加入第二终止液,得到具有结构式v的化合物。
优选的,在所述水解及闭环反应的所述步骤a中,所述第三有机溶剂包括丙酮、甲苯、甲醇、四氢呋喃和异丁醇中的至少一种;
所述第一保温反应的反应温度为所述第三有机溶剂的回流温度;
所述第一保温反应的反应时间为3~4h;
所述第一终止液包括碳酸钠溶液、醋酸钠溶液和氢氧化钾溶液中的至少一种。
优选的,所述水解及闭环反应中的所述步骤a还包括:
在向所得的第一反应液中加入第一终止液之后,对终止后的第一混合液进行后处理;
所述后处理的步骤包括:
(1)浓缩所述第一混合液中的有机溶剂,浓缩结束后依次加入甲苯、氯化钠与水进行萃取,静置分层;
(2)在上述步骤(1)所得的水相中加入甲苯继续萃取,静置分层;
(3)使用5%的氯化钠溶液洗涤上述步骤(2)所得的有机相,得到所述水解产物中间体。
优选的,在所述水解及闭环反应的所述步骤b中,所述碱性试剂为碱性固体和水的组合物或所述碱性固体,所述碱性固体包括氢氧化钾、叔丁醇钾、碳酸钠和氢氧化钠中的至少一种;
所述第四种溶剂包括甲苯、甲醇、丙酮、叔丁醇和异丙醇中的至少一种;
所述第二保温反应的反应温度为所述第四有机溶剂的回流温度;
所述第二保温反应的反应时间为2~3h;
所述第二终止液包括盐酸水溶液、醋酸水溶液和氯化铵水溶液中的至少一种。
优选的,所述水解及闭环反应的所述步骤b还包括:
在向所得的第二反应液中加入第二终止液之后,对终止后的第二混合液进行后处理;
所述后处理的步骤包括:
(1)使用洗涤剂洗涤所述第二混合液,所述洗涤剂包括氯化钠溶液、碳酸氢钠溶液和碳酸钠溶液中的至少一种;
(2)在上述步骤(1)所得的洗涤后的第二混合液中加入水相萃取剂,所述水相萃取剂包括甲苯、四氢呋喃和甲醇中的至少一种;
(3)浓缩上述步骤(2)所得的萃取相和所述第二混合液中的有机溶剂,浓缩结束后得到浓缩混合液,向所述浓缩混合液中加入溶剂,所述溶剂包括环己烷、石油醚、异丙醇、正己烷和甲醇中的一种或多种;
(4)对上述步骤(3)所得的加入所述溶剂的浓缩混合液进行冰水浴降温,以析出固体,搅拌时间为2~6h;
(5)对上述步骤(4)所得的混合物进行过滤抽干处理,对所得的固体进行干燥处理,得到所述具有结构式v的化合物固体,所述干燥处理的干燥温度为30℃~50℃以及干燥时间为2~4h。
优选的,所述水解及闭环反应中,所述具有结构式iv的化合物、所述碱性试剂之间的重量份数的比例为1:(0.20~0.30)。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
附图说明
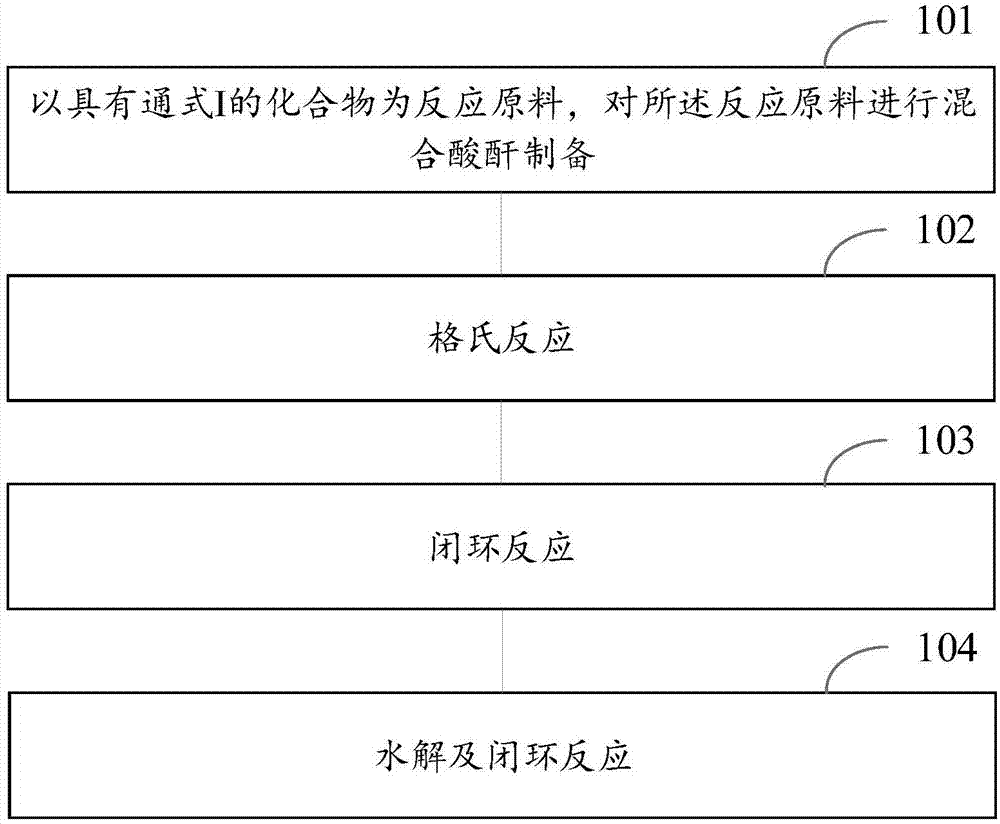
图1是本发明实施例的雌甾-4,9-二烯-3,17-二酮的制备方法流程图。
具体实施方式
以下结合具体实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。以下实施例中所采用的原材料和仪器均为市售。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法。图1是本发明实施例的雌甾-4,9-二烯-3,17-二酮的制备方法流程图,参考图1,所述雌甾-4,9-二烯-3,17-二酮的制备方法包括:
步骤101、以具有通式i的化合物为反应原料,对所述反应原料依次进行混合酸酐制备。
所述混合酸酐制备的反应过程可以包括以下步骤:
将使用第一有机溶剂溶解所述反应原料中,得到第一溶液,将所述第一溶液降温至第一预设温度并保温第一预设时长;
向所述第一溶液中缚酸剂和特戊酰氯,反应生成具有通式ii的化合物,反应结束后得到包含所述具有通式ii的化合物的第二溶液。
优选的,所述反应原料、所述缚酸剂和所述特戊酰氯之间的重量份数的比例为1:(0.72~1.62:):(1~1.1)。
所述第一有机溶剂可以包括四氢呋喃、无水乙醚和甲基四氢呋喃中的至少一种;所述缚酸剂可以包括吡啶、三乙胺和二苯甲酮中的至少一种。
优选的,所述第一预设温度为-30℃~-40℃;所述第一预设时长为2~4h。
步骤102、对步骤101所得的产品进行格氏反应。
所述格氏反应的反应过程可以包括以下步骤:
在氮气保护下,将包含所述具有通式ii的化合物的第二溶液降温至第二预设温度,滴加格氏试剂,滴加操作完成后进行保温反应,所述保温反应的反应时长为第二预设时长,所述保温反应结束后对所得的反应体系进行终止反应和后处理,得到包含具有结构式iii的化合物的第三溶液。
优选的,所述第二预设温度为-20℃~-60℃;所述第二预设时长为3~5h。
优选的,所述终止反应的反应过程可以包括以下步骤:向所述反应体系中加入反应终止液,所述反应终止液包括氯化铵水溶液、盐酸和醋酸水溶液中的至少一种。
步骤103、对步骤102所得的产品进行闭环反应。
所述闭环反应的反应过程可以包括以下步骤:
使用第二有机溶剂溶解所述具有结构式iii的化合物,向所得的溶液中加入碱性固体和水的组合物或所述碱性固体,进行闭环反应,待所述闭环反应结束后得到具有结构式iv的化合物。
优选的,所述闭环反应中,所述第二有机溶剂包括四氢呋喃、甲醇、异丙醇和甲苯中的至少一种;
所述碱性固体包括氢氧化钾、叔丁醇钾、碳酸钠和氢氧化钠中的至少一种。
优选的,所述闭环反应步骤中,所述闭环反应的反应温度为所述第二有机溶剂的回流温度;所述闭环反应的反应时间为10~12h。
优选的,所述闭环反应的反应过程还可以包括以下步骤:
对包含所述具有结构式iv的化合物的反应液进行静置分层,分离得到有机相和水相;
对所述有机相和所述水相进行后处理;
所述后处理的步骤可以包括:
(1)将洗液加入所述有机相中,洗去所述有机相中的杂质,所述洗液包括氯化钠溶液、碳酸氢钠溶液和碳酸钠溶液中的一种或多种;
(2)将萃取剂加入上述步骤(1)所得的所述水相中,用来萃取所述水相中残留的有机相,所述萃取剂包括甲苯、四氢呋喃和甲醇中的一种或多种;
(3)对上述步骤(2)所得的萃取相进行浓缩处理,以去除所述萃取相中的萃取剂;对所述有机相进行浓缩,以除去有机相中的有机溶剂。浓缩结束,合并萃取相和有机相的浓缩液,并加入环己烷、丙酮、正己烷和甲醇中的一种或多种溶剂;
(4)对上述步骤(3)所得的已加入所述溶剂的萃取相和有机相浓缩液进行冰水浴降温,以析出固体,搅拌时间为2~6h;
(5)将上述步骤(4)所得的冰水浴降温后的混合物进行过滤,并对所得的固体进行干燥,得到所述具有结构式iv的化合物固体,所述干燥的干燥温度为50℃~70℃以及干燥时间为2~4h。
优选的,所述闭环反应中,所述具有结构式iv的化合物与所述碱固体之间的重量份数的比例为1:(0.47~0.77)。
步骤104、对步骤103所得的产品进行水解及闭环反应,制得具有结构式v的化合物。
本发明实施例中,具有结构式v的化合物的合成路线如下:
优选的,所述水解及闭环反应的反应过程可以包括以下步骤:
步骤a、将所述具有结构式iv的化合物、盐酸和第三有机溶剂混合,进行第一保温反应,待所述第一保温反应结束后向所得的第一反应液中加入第一终止液,得到水解产物中间体;
步骤b、将所述水解产物中间体、碱性试剂和第四有机溶剂混合,进行第二保温反应,待所述第二保温反应结束后向所得的第二反应液中加入第二终止液,得到具有结构式v的化合物。
优选的,在所述水解及闭环反应的所述步骤a中,所述第三有机溶剂包括丙酮、甲苯、甲醇、四氢呋喃和异丁醇中的至少一种;
所述第一保温反应的反应温度为所述第三有机溶剂的回流温度;
所述第一保温反应的反应时间为3~4h;
所述第一终止液包括碳酸钠溶液、醋酸钠溶液和氢氧化钾溶液中的至少一种。
优选的,所述水解及闭环反应中的所述步骤a还可以包括以下步骤:
在向所得的第一反应液中加入第一终止液之后,对终止后的第一混合液进行后处理;
所述后处理的步骤可以包括以下步骤:
(1)浓缩所述第一混合液中的有机溶剂,浓缩结束后依次加入甲苯、氯化钠与水进行萃取,静置分层;
(2)在上述步骤(1)所得的水相中加入甲苯继续萃取,静置分层;
(3)使用5%的氯化钠溶液洗涤上述步骤(2)所得的有机相,得到所述水解产物中间体。
优选的,在所述水解及闭环反应的所述步骤b中,所述碱性试剂为碱性固体和水的组合物或所述碱性固体,所述碱性固体包括氢氧化钾、叔丁醇钾、碳酸钠和氢氧化钠中的至少一种;
所述第四种溶剂包括甲苯、甲醇、丙酮、叔丁醇和异丙醇中的至少一种;
所述第二保温反应的反应温度为所述第四有机溶剂的回流温度;
所述第二保温反应的反应时间为2~3h;
所述第二终止液包括盐酸水溶液、醋酸水溶液和氯化铵水溶液中的至少一种。
优选的,所述水解及闭环反应的所述步骤b还可以包括以下步骤:
在向所得的第二反应液中加入第二终止液之后,对终止后的第二混合液进行后处理;
所述后处理的步骤可以包括:
(1)使用洗涤剂洗涤所述第二混合液,所述洗涤剂包括氯化钠溶液、碳酸氢钠溶液和碳酸钠溶液中的至少一种;
(2)在上述步骤(1)所得的洗涤后的第二混合液中加入水相萃取剂,所述水相萃取剂包括甲苯、四氢呋喃和甲醇中的至少一种;
(3)浓缩上述步骤(2)所得的萃取相和所述第二混合液中的有机溶剂,浓缩结束后得到浓缩混合液,向所述浓缩混合液中加入溶剂,所述溶剂包括环己烷、石油醚、异丙醇、正己烷和甲醇中的一种或多种;
(4)对上述步骤(3)所得的加入所述溶剂的浓缩混合液进行冰水浴降温,以析出固体,搅拌时间为2~6h;
(5)对上述步骤(4)所得的混合物进行过滤抽干处理,对所得的固体进行干燥处理,得到所述具有结构式v的化合物固体,所述干燥处理的干燥温度为30℃~50℃以及干燥时间为2~4h。
优选的,所述水解及闭环反应中,所述具有结构式iv的化合物、所述碱性试剂之间的重量份数的比例为1:(0.20~0.30)。
本发明实施例提供的雌甾-4,9-二烯-3,17-二酮的制备方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
下面通过以下实施例对本申请提供的雌甾-4,9-二烯-3,17-二酮的制备方法进行详细说明。
实施例1
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-30℃,滴加50g特戊酰氯,0.5h滴完后,滴入45g三乙胺,1h滴完后,保温反应2h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物的第二溶液,第二溶液的体积约为450ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,加入450ml的第二溶液,温度降至-40℃,滴加300ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应3h,液相检测结果指示反应完全后,加入200g质量分数为1%的醋酸水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物的第三溶液,第三溶液的体积约为850ml。
闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的该四口烧瓶中,依次加入850ml第三溶液和50g氢氧化钾,搅拌溶解并升温至回流,保温反应10h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的nacl溶液,洗去有机相杂质。向水相中加入100ml四氢呋喃,萃取,后合并有机相。减压浓缩四氢呋喃有机层至浓不出为止。加环己烷,冰水浴降温,搅拌5h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv的化合物,所述具有结构式iv的化合物的重量为65.1g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和2g碳酸钠调ph至中性。减压浓缩尽丙酮,加入800ml甲苯、10g氯化钠和200ml水并搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤合并后的甲苯层,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加100g异丙醇和25g叔丁醇钾固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加醋酸水溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,减压浓缩甲苯有机层至浓不出为止。加环己烷,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为67.1g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
实施例2
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-35℃,同时滴加50g特戊酰氯和45g三乙胺,1h滴完后,保温反应2h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物的第二溶液,第二溶液的体积约为450ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,加入450ml的第二溶液,温度降至-45℃,滴加300ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应3h,液相检测结果指示反应完全后,加入200g质量分数为1%的氯化铵水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物第三溶液,第三溶液的体积约850ml,减压浓缩四氢呋喃有机层至浓不出为止。
闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的该四口烧瓶中,依次加入含化合物iii的第三溶液,500ml甲苯和30g氢氧化钾,搅拌溶解并升温至回流,保温反应12h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的nacl溶液,洗去有机相杂质。向水相中加入50ml甲苯,萃取,后合并有机相。减压浓缩甲苯有机层至浓不出为止。加环己烷和丙酮,冰水浴降温,搅拌3h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv的化合物,所述具有结构式iv的化合物的重量为63.5g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和2g碳酸钠调ph至中性。减压浓缩尽丙酮,加入800ml甲苯、10g氯化钠和200ml水并搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤合并后的甲苯层,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加140g叔丁醇和20g叔丁醇钾固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加盐酸水溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,水相用100ml甲苯萃取,合并甲苯有机相。减压浓缩甲苯有机层至浓不出为止。加石油醚和异丙醇,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为65.3g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
实施例3
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-30℃,同时滴加50g特戊酰氯和45g三乙胺,1h滴完后,保温反应2h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物的第二溶液,第二溶液的体积约为450ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,加入450ml的第二溶液,温度降至-40℃,滴加300ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应4h,液相检测结果指示反应完全后,加入200g质量分数为1%的氯化铵水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物的第三溶液,第三溶液的体积约为850ml,减压浓缩四氢呋喃有机层至浓不出为止。
闭环反应
选取洁净烦躁的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的该四口烧瓶中,依次加入含化合物iii的第三溶液、500ml甲苯和30g氢氧化钾,搅拌溶解并升温至回流,保温反应12h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的nacl溶液,洗去有机相杂质。向水相中加入50ml甲苯,萃取,后合并有机相。减压浓缩甲苯有机层至浓不出为止。加环己烷和丙酮,冰水浴降温,搅拌3h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv的化合物,所述具有结构式iv的化合物的重量为64.3g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和2g碳酸钠调ph至中性。减压浓缩尽丙酮,加入甲苯800ml、10g氯化钠和200ml水搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加200g叔丁醇和30g叔丁醇钾固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加盐酸水溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,水相用100ml甲苯萃取,合并甲苯有机相。减压浓缩甲苯有机层至浓不出为止。加正己烷和甲醇,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为66.7g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
实施例4
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-40℃,滴加55g特戊酰氯,0.5h滴完后,滴入36g吡啶,1h滴完后,保温反应4h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物的第二溶液,第二溶液的体积约约450ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,加入450ml的第二溶液,温度降至-25℃,滴加300ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应3h,液相检测结果指示反应完全后,加入200g质量分数为1%的醋酸水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物的第三溶液,第三溶液的体积约为850ml。
闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的该四口烧瓶中,依次加入850ml第三溶液和40g氢氧化钾,搅拌溶解并升温至回流,保温反应11h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的碳酸氢钠溶液,洗去有机相杂质。向水相中加入50ml甲苯,萃取,后合并有机相。减压浓缩四氢呋喃有机层至浓不出为止。加环己烷,冰水浴降温,搅拌4h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv的化合物,所述具有结构式iv的化合物的重量为64.5g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml物质的量浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和1.55g醋酸钠调ph至中性。减压浓缩尽丙酮,加入甲苯800ml、10g氯化钠和200ml水搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加160g叔丁醇和25g氢氧化钾固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加盐酸水溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,减压浓缩甲苯有机层至浓不出为止。加正己烷和甲醇,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为66.9g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大等优点。
实施例5
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-35℃,同时滴入55g特戊酰氯和81g二苯甲酮,1.5h滴完后,保温反应3h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物的第二溶液,第二溶液的体积约为500ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,加入500ml的第二溶液,温度降至-35℃,滴加330ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应3h,液相检测结果指示反应完全后,加入220g质量分数为1%的醋酸水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物iii的第三溶液,第三溶液的体积约为900ml,减压浓缩四氢呋喃有机层至浓不出为止。
闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的该四口烧瓶中,依次加入含化合物iii的第三溶液、500ml甲苯、45g氢氧化钾,搅拌溶解并升温至回流,保温反应11h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的碳酸钠溶液,洗去有机相杂质。向水相中加入50ml甲苯,萃取,后合并有机相。减压浓缩四氢呋喃有机层至浓不出为止。加环己烷,冰水浴降温,搅拌4h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv化合物,所述具有结构式iv的化合物的重量为62.5g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和1g氢氧化钾调ph至中性。减压浓缩尽丙酮,加入甲苯800ml、10g氯化钠和200ml水搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加100g异丙醇和25g氢氧化钠固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加氯化铵溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,减压浓缩甲苯有机层至浓不出为止。加环己烷,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为64.2g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生产等优点。
实施例6
混合酸酐的制备
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的装配温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中,依次加入400ml四氢呋喃和50g具有结构式i的化合物,搅拌溶解并降温至-45℃,滴加50g特戊酰氯,0.5h滴完后,滴入45g三乙胺,1h滴完后,保温反应2h。液相检测结果指示反应完全后,得到包含具有通式ii的化合物ii的第二溶液,第二溶液的体积约为450ml。
格氏反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向选取的处于机械搅拌状态的四口烧瓶中装,加450ml的第二溶液,温度降至-45℃,滴加300ml浓度为1.1mol/l的格氏试剂(氯[3-(2,5,5-三甲基-1,3-二恶烷-2-基)丙基]-镁),2~3h滴完,保温反应3h,液相检测结果指示反应完全,加入200g质量分数为1%的醋酸水溶液,进行终止反应。将反应液转移至分液漏斗中,分层;水相用100ml四氢呋喃萃取,再分层;合并有机相,得到包含具有结构式iii的化合物iii的第三溶液,第三溶液的体积约为850ml。
闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
在氮气保护下,向处于机械搅拌状态的四口烧瓶中,依次加入850ml第三溶液和50g氢氧化钠,搅拌溶解并升温至回流,保温反应10h,液相检测结果指示反应完全。
将所得的反应液转移至分液漏斗中,静置分层。向有机层加入200ml质量分数为10%的nacl溶液,洗去有机相杂质。向水相中加入100ml四氢呋喃,萃取,后合并有机相。减压浓缩四氢呋喃有机层至浓不出为止。加异丙醇和石油醚,冰水浴降温,搅拌4h,得到大量白色固体,过滤抽干后,60℃干燥3h,得到具有结构式iv的化合物,所述具有结构式iv的化合物的重量为65.5g。
水解及闭环反应
选取洁净干燥的1000ml四口烧瓶,该四口烧瓶装配有洁净干燥的温度计和恒压滴液漏斗。
向处于机械搅拌状态的四口烧瓶中,依次加入100g具有结构式iv的化合物、500ml丙酮、25ml浓度为2mol/l的hcl水溶液,搅拌溶解并升温至回流,保温反应3h,液相检测结果指示反应完全。
加入150ml水和1g氢氧化钾调ph至中性。减压浓缩尽丙酮,加入800ml甲苯、10g氯化钠和200ml水搅拌,静置分层。水相用100ml甲苯萃取,合并甲苯层,用100g质量分数为5%的氯化钠溶液洗涤,再分层,舍弃水相,得含有水解中间体的有机相溶液。
向含有水解中间体的甲苯溶液中加160g叔丁醇和25g叔丁醇钾固体,搅拌溶解并升温至回流,保温反应2h,液相检测结果指示反应完全。加醋酸水溶液终止反应,调ph至中性。静置分层,有机相用300g质量分数为30%的氯化钠溶液洗涤,减压浓缩甲苯有机层至浓不出为止。加环己烷,冰水浴降温,搅拌3h,得到大量淡黄色固体,过滤抽干后,40℃干燥3h,得到具有结构式v的化合物,具有结构式v的化合物的质量为67.8g。
本发明实施例提供了一种雌甾-4,9-二烯-3,17-二酮的制备方法,该方法具有合成过程中操作步骤简单、原料便宜、有机溶剂用量小、后处理容易和易于放大生成等优点。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例。凡属于本发明思路下的技术方案均属于本发明的保护范围。应该指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下的改进和润饰,这些改进和润饰也应视为本发明的保护范围。
以上对本发明所提供的一种雌甾-4,9-二烯-3,17-二酮的制备方法的应用进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
- 还没有人留言评论。精彩留言会获得点赞!