一种海洋真菌来源的醌类化合物及其制备方法与应用与流程
本发明涉及药物化合物的技术领域,更具体地,涉及一种海洋真菌来源的醌类化合物及其制备方法与应用。
背景技术:
炎症是具有血管系统的活体组织对各种损伤因子的刺激所产生的防御反应,典型的反应是出现红、肿、热、痛等临床症状,是机体对内外环境中有害刺激所产生的一种复杂的生理和病理反应。炎症反应是一种保护性防御反应,又是引起人类多种重大疾病的共同通路,参与人体感染、肿瘤、心脑血管病、老年痴呆和神经退行性疾病、变态反应疾病、精神病等许多重大疾的发生和发展过程。临床上,抗炎药物是仅次于抗感染药物的第二大类药物。因此,寻找新型、高效的抗炎药物一直是科研人员的研究热点。
海洋生物在特殊环境之中,已发展出独特的代谢方式,很多文献已证明海洋生物来源的真菌能够产生结构新颖、生理活性显著的各类次级代谢产物。其代谢产物具有抗菌、抗肿瘤、免疫调节、酶抑制等多种药用价值。目前,从包括海洋真菌在内的海洋微生物中寻找新的药源已成为国际国内研究的热点。
技术实现要素:
本发明的目的在于提供一种能够有效抗炎的新型化合物,本发明从冠突散囊菌eurotiumcristatum33yh-wz-18的发酵产物中分离得到一种新型化合物,发明人通过研究发现,该新型化合物具有抗炎活性,可应用于制备抗炎药物。
本发明还的另一个目的是提供所述新型化合物的制备方法。
本发明的上述目的是通过以下技术方案给予实现的:
一种海洋真菌来源的醌类化合物,其结构式如式(i)所示:
所述醌类化合物的制备方法,包括如下步骤:
s1.将真菌eurotiumcristatum33yh-wz-18接入种子培养基,摇床培养,得到种子培养液;
s2.将种子培养液接入发酵培养基中,静置培养得到发酵物;
s3.将所述发酵物先分离菌体和菌液,用甲醇浸泡菌体,减压浓缩得到菌体粗体物,菌液用乙酸乙酯萃取,减压浓缩后得菌液粗体物,将菌体和菌液粗体物进行合并获得粗浸膏;分别以正己烷、乙酸乙酯、甲醇作为洗脱剂,对粗浸膏采用减压柱进行粗分,分别得到正己烷相、乙酸乙酯相、甲醇相,再对正己烷相进行分离纯化,得到式(i)化合物;
其中,所述真菌为冠突散囊菌eurotiumcristatum33yh-wz-18。所述真菌eurotiumcristatum33yh-wz-18已在广东省微生物菌种保藏中心保藏,保藏地址为广州市先烈中路100号大院59号楼5楼,保藏日期是2018年7月18日,保藏号是gdmccno:60420。
本发明所述海洋真菌eurotiumcristatum33yh-wz-18菌株的来源为一只采自广东湛江徐闻角尾乡的艳红海百合,从其腕足上分离得到的真菌,其分类命名为eurotiumcristatum33yh-wz-18。
优选地,步骤s1中,所述种子培养基为pdb液体培养基。所述pdb液体培养基可参考现有的pdb液体培养基条件制备,包括但不限于以下方法,所述pdb液体培养基按照每升水中加入30g海盐和24gpdb培养基粉末进行配制。
优选地,步骤s1中,所述摇床培养的条件是:25℃下,摇床转速100~150rpm,培养时间为3~5天。
优选地,步骤s2中,所述发酵培养基与种子培养基相同。
优选地,步骤s2中,所述静置培养的条件是:静置培养的时间为30天,静置培养的温度为室温。
优选地,步骤s3中,所述正己烷相用硅胶柱进行层析分离,硅胶柱进行层析分离时分别用10:0、9:1、8:2、7:3、6:4、5:5、4:6、3:7、2:8、1:9、0:10的石油醚-乙酸乙酯进行梯度洗脱;对所述8:2、7:3的石油醚-乙酸乙酯洗脱部分合并,用凝胶柱色谱和高效液相纯化,得到式(i)化合物。
所述硅胶柱为本领域所用到的常规硅胶柱,硅胶柱的目数为200~300目。
优选地,所述凝胶柱色谱的条件为,凝胶柱为sephadexlh-20,以甲醇为洗脱剂;高效液相色谱的条件是:流动相:80%meoh-h2o;流速:1ml/min,色谱柱:半制备柱ultimatexb-c18,10×250mm,5μm;仪器essentialc-16。
经现有的抗炎活性测试发现,本发明所述醌类化合物具有抗炎活性,可用于制备抗炎药物,因此,其在制备抗炎药物中的应用,都应在本发明的保护范围之内。
与现有技术相比,本发明的有益效果是:
本发明从冠突散囊菌eurotiumcristatum33yh-wz-18的发酵产物中分离得到一种新型化合物,该新型化合物具有抗炎活性,可应用于制备抗炎药物,具有广阔的应用前景广阔。另外制备方法简单,成本低廉。
附图说明
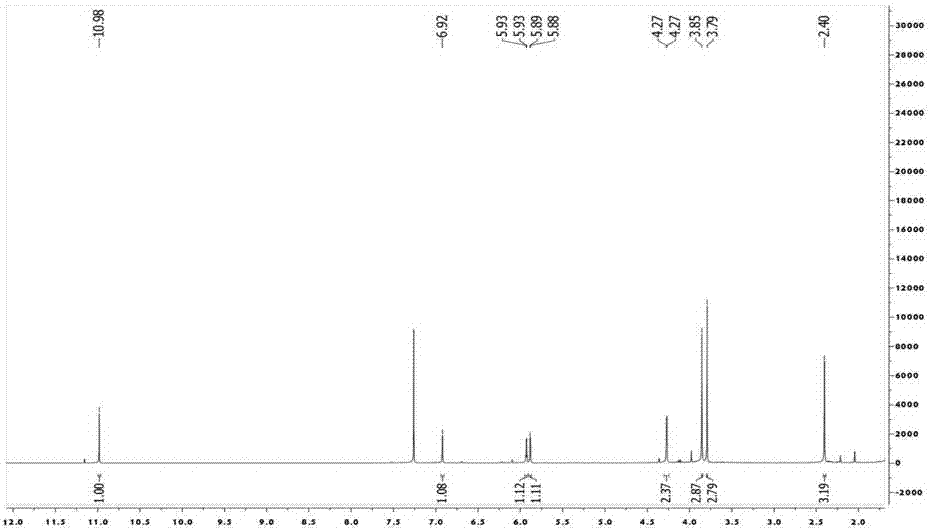
图1为本发明实施例1所得化合物的核磁共振氢谱。
图2为本发明实施例1所得化合物的核磁共振碳谱。
图3为本发明实施例1所得化合物的hresims质谱。
具体实施方式
下面结合具体说明书附图和实施例对本发明作进一步的解释说明,但具体实施例并不对本发明作任何限定。除非特别说明,实施例中所涉及的试剂、方法均为本领域常用的试剂和方法。
实施例1化合物的提取与表征
1、化合物的具体制备步骤如下:
s1.种子培养液的获得
s11.配制种子培养基:
该种子培养基为pdb液体培养基,pdb液体培养基按照3%的粗海盐、每升24克pdb进行配制。平均分配到4个1l锥形瓶内,经高温灭锅121℃(0.1mpa)条件灭菌25min,之后冷却至室温,静置24小时。
s12.种子的培养:将真菌eurotiumcristatum33yh-wz-18接入种子培养基,将接种后的锥形瓶放置在摇床上25℃恒温培养72小时,获得种子培养液。
s2.发酵培养:采用pdb液体培养基,置于1l锥形瓶混匀后密封,经121℃(0.1mpa)高温灭菌25min后冷却至室温放置2天,挑选瓶内培养基无染菌现象的进行接种,每瓶接种10ml菌种(种子培养液),共接种104瓶,静置培养30天得发酵物。
s3.提取分离:
发酵培养后,将发酵物先分离菌体和菌液,用甲醇浸泡菌体,减压浓缩得到菌体粗体物,菌液用乙酸乙酯萃取,减压浓缩后得菌液粗体物,将菌体和菌液粗体物进行合并获得粗浸膏,分别以正己烷、乙酸乙酯、甲醇作为洗脱剂,对粗浸膏采用减压柱进行粗分,分别得到正己烷相、乙酸乙酯相、甲醇相。
所得正己烷相用硅胶柱进行层析分离,硅胶柱进行层析分离时分别用10:0、9:1、8:2、7:3、6:4、5:5、4:6、3:7、2:8、1:9、0:10的石油醚-乙酸乙酯进行梯度洗脱,所得8:2、7:3的石油醚-乙酸乙酯洗脱部分合并,用凝胶柱色谱和高效液相纯化,凝胶柱色谱的条件为:凝胶柱为sephadexlh-20,以甲醇为洗脱剂。高效液相色谱的条件是:流动相:80%meoh-h2o;流速:1ml/min,色谱柱:半制备柱ultimatexb-c18,10×250mm,5μm;仪器essentialc-16,得到黄色固体。
2、表征
对该黄色固体进行核磁共振检测,所得谱图如图1~3所示,经分析检测,该化合物结构的理化性质数据如下:
uv(meoh)λmax(logε)205(3.32),264(2.90)nm;
ir(neat)νmax2959,2920,2851,1650,1602,1444,1260,1238,1094,1029,799cm-1;
1hand13cnmrdata(cdcl3,400and100mhz),见表1;
表1
hr-esimsm/s349.0486[m-h]-(calcdforc17h14o6cl,349.0484),详细信息如下表所示:
从质谱和核磁共振的结果分析可确定化合物的分子式为c17h15o6cl,其结构式如下:
实施例2化合物的抗炎活性测试
1、实验材料
吲哚美辛(购自于源叶生物);脂多糖(购自于索莱宝);no试剂盒(购自于碧云天)。
2、实验方法
采用上述所得样品作为实验对象,以吲哚美辛为阳性对照,所得样品与吲哚美辛均用dmso配制初始浓度50mm的待测样液(所得样品待测样液、吲哚美辛待测样液)。
操作步骤:
s1.raw264.7(小鼠单核巨噬细胞)接种于96孔板中(浓度为1×105个/孔),孵化12h。
s2.弃去旧培养液,将lps(脂多糖)(1μg/ml)与待测样液混合,用新鲜培养液稀释成相应浓度,分别加入96孔板中,作用24h。
s3.吸取上清液50μl到新的96孔板中,每孔分别加入50μl的no试剂ⅰ和no试剂ⅱ(碧云天),随后在酶标仪中于540nm测其od值。
抗炎活性结果:
经测试计算,该醌类化合物的测试结果为ic50=1.48±0.84μm,其抑制率计算公式为[od(模型组)-od(加药组)]/[od(模型组)-od(正常组)]×100%。
其中,od(模型组)中的模型组指的是lps诱导的raw264.7细胞实验组。
od(加药组)中的加药组指的是加入目标化合物样品的lps诱导的raw264.7细胞实验组。
od(正常组)中的正常组指的是只有raw264.7细胞的实验组。
相对于阳性对照(吲哚美辛,ic50=41.0±1.0μm),该醌类化合物具有良好抗炎活性。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!