一种外源表达PEPR1蛋白的方法与流程
本发明涉及基因工程技术领域,具体地,涉及一种外源表达pepr1蛋白的方法。
背景技术:
pepr1是模式植物拟南芥中的一个富亮氨酸重复类受体蛋白激酶(leucine-richrepeatsreceptorkinase,lrr-rk),lrr-rk家族分为16个亚家族,是一类研究最为深入的受体蛋白激酶,其组成包括一个富亮氨酸重复类的胞外识别结构域,单次跨膜区,胞内激酶结构域。pepr1蛋白属于lrr-rk第六家族成员,在pepr1的胞外区有27个lrr单元,n端和c端各有一对半胱氨酸形成“帽子”结构来稳定胞外区的疏水核心。
pepr1受体蛋白激酶识别的是由植物体自身分泌的一类内源性多肽分子,叫做损伤分子相关模式,当植物体出现伤口或者是受损伤时受刺激后分泌的,识别后可以激活胞内激酶活性,引发植物的活性氧爆发、胼胝质沉积等一系列免疫反应,帮助植物抵抗病菌的入侵。体外获得pepr1蛋白,可以用于筛选新型的小分子药物,筛选出可以激活植物免疫反应的小分子药物,当病菌入侵时,植物可以抵抗病菌的侵害,研制新型抗病菌药物,促进植物的抗病表型并提高植物的产量。
目前的研究内容主要集中在探究pepr1这类受体蛋白的胞外识别结构域是如何识别胞外的信号源识别后被激活的机制。主要手段是通过x射线衍射技术获取蛋白的精细三维结构,在分子水平上阐述上述问题。由于pepr1蛋白只有一个疏水的跨膜区,这对于在体外重组该全长蛋白是一个很大的阻碍。由于蛋白的疏水跨膜区是将细胞膜上的蛋白牢牢锚定在细胞膜上的区域,跨膜区越多,蛋白在细胞膜上越稳定,若跨膜区较多的膜蛋白在体外重组过程中可以采用去垢剂等化学试剂来模拟细胞膜,进而成功获得膜蛋白的体外成功表达,但是对于只有一个疏水跨膜区的蛋白,比如pepr1蛋白,带有跨膜区的全长蛋白在体外表达过程中极不稳定,因此会导致蛋白在体外不表达或者是形成构象不正确的蛋白。因此单次疏水跨膜区极其不利于蛋白在细胞膜上的稳定性,目前可采取的策略是只将pepr1蛋白的胞外区域进行体外扩增并构建到适当的表达载体上,然后获得其稳定构象的胞外结构域的蛋白。
蛋白质大分子在生物体内承担着多种多样的生理功能,研究蛋白质的结构和功能对于人类了解生理调控、机体代谢具有重要意义,但往往需要在体外获取具有正确构象及生理功能的重组蛋白,才能保证一系列研究的顺利开展和实施成功。原核表达系统很大程度地改善了重组外源蛋白表达量不高以及下游分离加工难度大等问题。其中融合蛋白表达技术的表达模式是:原核启动子→sd序列→起始密码子→原核结构基因片断→目的基因序列→终止密码子,是比较常用的表达载体,尤其适用于小分子多肽和蛋白的表达。然而由于pepr1蛋白为大分子真核胞外分泌蛋白,采用原核表达系统外源表达pepr1蛋白往往形成包涵体,不能获得构象正确的蛋白。现有技术采用真核表达系统来生产pepr1蛋白,但表达量都不高,通常在0.09mg/l。这样的表达量远远不能满足后续产业对pepr1蛋白的需求。因此,急需一种能够提高pepr1蛋白表达量、成本低廉的方法以弥补现有技术的不足。
技术实现要素:
本发明的一个目的是提供一种外源表达pepr1蛋白的方法,该方法能够提高pepr1蛋白的表达量,并且得到的pepr1蛋白的构象正确,符合预期。
本发明的另一个目的是提供一种外源表达pepr1蛋白的表达载体。
本发明采用的主要技术方案为:考虑到pepr1胞外编码区不存在内含子区域,本发明从拟南芥基因组中直接扩增出的pepr1的胞外结构域。对pepr1基因序列进行结构域分析,采用信号肽预测网站(signalp)和疏水跨膜区预测网站(tmhmm)预测出pepr1的疏水跨膜区和信号肽区域。在疏水跨膜区前的这一段区域即为胞外区。考虑到pepr1蛋白属于分泌蛋白,其本身的n端信号肽会带领着合成好的肽链在内膜系统中进行后期修饰和加工,并且引领加工好的蛋白分泌到细胞膜外,当信号肽的功能完成后,信号肽会在后期被剪切掉,成为成熟的分泌蛋白。通过软件预测后初步确定pepr1跨膜区为770-792aa。跨膜区前端边界处可以前后3-4个氨基酸多做几个片段长度,以防预测位置与实际切割位置有误差造成蛋白错误折叠。最终确定的pepr1胞外区长度是1-767aa,其氨基酸序列如seqidno.1所示。
限制性内切酶的选择是建立在目的基因序列中不能够出现所挑选的位于基因两侧的这两种限制性内切酶的酶切序列,否则在进行后期的酶切实验过程中,所要连入载体的基因序列会被限制性内切酶所切割,从而无法得到成功的重组质粒。基于上述原则,本发明在筛选酶切位点时,选择了两对限制性内切酶bamh1和xho1,bgl2和sal1。bamh1和bgl2互为同尾酶,xho1和sal1互为同尾酶。这两组同尾酶对目的基因进行酶切,每一组同尾酶中各选取一个,两两搭配进行使用。共有四种组合方式:bamh1和xho1,bamh1和sal1,bgl2和xho1,bgl2和sal1。同时本发明发现在pepr1基因中没有bamh1和sal1这两种限制性内切酶对应的酶切位点。因此选择bamh1和sal1这个组合进行酶切。前述四种组合的合理运用可以最大限度地规避目的基因中出现酶切位点序列,减少后期采用点突变来消除酶切位点的工作量。
外源载体为载体pfastbactmhem采用bamh1和xho1这两种限制性内切酶进行处理后备用。而目的基因两端的酶切位点就在bamh1&xho1,bamh1&sal1,bgl2&xho1,bgl2&sal1这四种组合方式中选择,这样目的片段才能顺利连入已切割的载体。pepr1基因序列中不存在bamh1和sal1这两种限制性内切酶的酶切位点,因此选用bamh1和sal1作为基因两端的酶切位点,而不是“bamh1和xho1”、“bgl2和xho1”或“bgl2和sal1”。当后期采用bamh1和sal1这两种限制性内切酶对pepr1进行切割时,pepr1基因不会被这两种酶所切断。然后根据密码子的容错性原理将碱基进行点突变,这样既消除该酶切位点,同时也并不会造成氨基酸的突变。选择了酶切位点后,在(酶切位点的)5’和3’端再分别加上合适的保护碱基即可。本发明pepr1基因的外显子如seqidno.2所示。
在外源载体的构建方面,发明人充分考虑内切酶的选择原则和外源表达载体信号肽的信号传输功能,经过酶切位点的筛选和在表达载体中引入杆状病毒来源的抑血细胞聚集素hemolin信号肽替换pepr1蛋白自身的信号肽。pepr1蛋白自身的信号肽是其序列n端的前28个氨基酸。本发明构建的外源表达载体上与目的基因n端连接的前端加入一段杆状病毒来源的抑血细胞聚集素hemolin信号肽。
进一步地,所述外源载体为载体pfastbactmhem,其是在载体pfastbactm1的bamh1酶切位点前加入hemolin信号肽得到,所述hemolin信号肽的核苷酸序列如seqidno.3所示。
信号肽在蛋白表达加工过程中的重要作用,通常是将pepr1这类植物蛋白在昆虫细胞中进行外源表达。由于无法得知植物蛋白自身的信号肽转入到昆虫细胞中是否依然可以正常行使自身的功能,故本发明的一个优选的实施例中,将昆虫细胞表达系统中的杆状病毒表达载体pfastbac-tm1进行了改造,新改造好的载体为pfastbactm-hem。即在pfastbac-tm1载体上与目的基因n端连接的前端部分上加入一段杆状病毒来源的抑血细胞聚集素(hemolinpeptide)信号肽。本发明意外发现采用hem信号肽比采用pepr1自身的n端信号肽效果要更好,可以被昆虫体系识别、剪切并引导穿越内质网膜的序列作为分泌信号肽。同时本发明在目的基因的c末端加上便于纯化的his标签,采用在3’-5’端引物上加上6个组氨酸密码子序列和终止密码子序列,使阅读框一直向下读取直至到读取完his标签的密码子,然后读取完终止密码子。翻译结束。
选用pfastbactm-hem这一本发明新改造的载体作为表达载体,需要将连入的目的基因片段(pepr1外显子基因)前的自身信号肽去掉,因此连入的目的片段是pepr1-29-767,去掉了n端的28个氨基酸。本发明的pepr1蛋白是选用pfastbactm-hem载体加上pepr1-29-767基因片段,能够获得正确折叠表达的pepr1胞外区蛋白(氨基酸序列如seqidno.1所示),并提高pepr1的表达量。
基于本发明的整体技术构思与技术发明点剖析,本发明提供了一种外源表达pepr1蛋白的方法,包括步骤:
(1)构建含有杆状病毒来源的抑血细胞聚集素hemolin信号肽的载体;
(2)克隆得到pepr1外显子基因,将pepr1外显子基因连入步骤(1)的载体上,得到外源表达载体;
(3)外源表达载体转染昆虫细胞,获得重组杆状病毒,病毒侵染昆虫细胞,培养该昆虫细胞后,收获细胞和上清,上清液经层析,纯化后获得构象正确的pepr1蛋白。
上述方法中,步骤(1)所述载体的构建方法是在pfastbactm1的bamh1酶切位点前加入hemolin信号肽得到pfastbactmhem载体;所述hemolin信号肽的核苷酸序列如seqidno.3所示。
本发明上述方法所述pepr1蛋白的氨基酸序列如seqidno.1所示。
本发明上述方法所述pepr1外显子基因的核苷酸序列如seqidno.2所示。
本发明提供的外源表达pepr1蛋白方法中,步骤(2)的pepr1外显子基因连入载体前采用bamh1和sal1酶切pepr1外显子基因。
进一步地,步骤(2)中,采用bamh1和xho1酶切步骤(1)构建得到的载体,酶切后与pepr1外显子基因连接,构建外源表达载体。
本发明提供的外源表达pepr1蛋白方法中,另一个关键步骤在于采用expressionsystemesfaf培养基扩大培养昆虫细胞后,再用步骤(3)转染昆虫细胞后获得的重组杆状病毒侵染前述扩大培养的昆虫细胞,这样能够获得更多量的构象正确的pepr1蛋白。
本发明采用sf900ii培养基和expressionsystemesfaf培养基分别进行同样操作的昆虫细胞扩大培养后,同样数量级的昆虫细胞(hi5细胞或sf21细胞系),expressionsystemesfaf培养基的实验组中构象正确的pepr1蛋白显著提高,排除了是由于hi5细胞总量变多而导致pepr1蛋白变多的可能。
在本发明的优选实施例中,本发明方法的步骤(3)中,采用义翘神州公司的expressionsystemesfaf培养基将昆虫细胞稀释至细胞浓度为0.3×106-1×106个/ml,过夜培养,当生长到1.6×106-2.0×106个/ml时,加入体积比1:50的含pepr1的重组杆状病毒,培养后收集细胞培养液上清。
本发明提供了上述方法制备得到的pepr1蛋白。
另一方面,本发明提供了一种表达pepr1蛋白的外源表达载体,该载体上与目的基因n端连接的前端加入一段杆状病毒来源的抑血细胞聚集素hemolin信号肽。
优选地,上述外源表达载体为载体pfastbactmhem,是在载体pfastbactm1的bamh1酶切位点前加入hemolin信号肽得到,所述hemolin信号肽的核苷酸序列如seqidno.3所示。
进一步地,本发明提供了上述外源表达pepr1蛋白的方法、该方法所得到的pepr1蛋白、上述外源表达载体在筛选可以激活植物免疫反应的小分子药物、研制植物抗菌药、提高植物抗病表型或提高植物产量中的应用。
基于上述技术方案,本发明取得了以下有益效果:
(1)本发明方法解决了长期以来使用大肠杆菌原核表达体系一直无法正确表达pepr1蛋白的技术难题。由于pepr1蛋白属于分泌类的植物蛋白,在蛋白折叠、分泌的过程中需要分子内二硫键的正确形成以及后期蛋白质表面的糖基化修饰,但是在原核表达系统中缺少糖基化修饰这一过程并且容易导致二硫键的错配。本发明方法首次采用hi5和sf21细胞系对植物拟南芥中的富亮氨酸重复类受体蛋白pepr1进行体外重组表达。
(2)在质粒构建环节采用同尾酶两两组合,发现在pepr1基因中没有bamh1和sal1这两种限制性内切酶对应的酶切位点,因此选择bamh1和sal1这个组合进行酶切。限制性内切酶合理运用可以最大限度地规避目的基因中出现酶切位点序列,减少后期采用点突变来消除酶切位点的工作量,大大提高载体的利用率和构建效率。
(3)由于不同的目的蛋白外源形成的过程千差万别的,外源表达的方法也各不相同,所用的表达载体,信号肽,培养基,培养温度都能影响蛋白的表达量,本发明通过改造载体,选择适用于细胞分泌表达的信号肽,获得了更适合pepr1蛋白表达的载体pfastbactmhem,通过选择适宜的培养基种类expressionsystemesfaf培养基,有助于外源蛋白的成功分泌和表达,提高获得正确的重组蛋白的得率。
(4)本发明在小量表达检测阶段采用非还原性(无dtt)检测可以在凝胶过滤层析之前就分析出正确构象的蛋白分子和聚集体之间的比例关系,大大地节省成本和时间。
附图说明
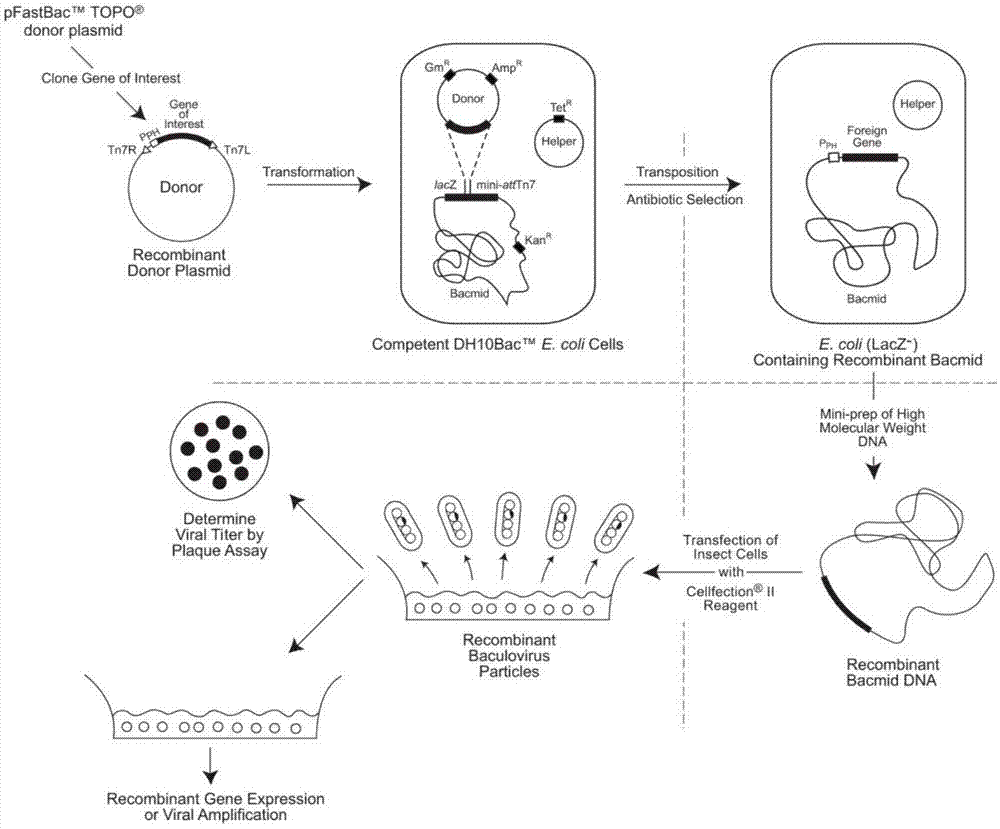
图1为本发明所采用的bac-to-bac杆状病毒表达系统的过程示意图。
图2为pepr1基因的pcr扩增产物凝胶电泳图,利用seqidno.4-5所示引物扩增出的pepr1基因大小约为2217bp,图中显示基因片段位于2000-3000bp之间。
图3为pfastbactm1杆状病毒载体图谱及多克隆位点图示。
图4a为pfastbactmhem杆状病毒载体图谱,pfastbactmhem杆状病毒载体是在载体pfastbactm1的bamh1酶切位点前加入hemolin信号肽得到,所述hemolin信号肽的核苷酸序列如seqidno.3所示;图4b为pfastbactmhem的多克隆位点,限制性酶切位点如图所示代表实际的切割位点,下划线标记潜在的终止密码子。构建的信号肽从atg开始翻译,翻译出hemolin的信号肽直接与外源表达的目的基因n端融合。
图5为expressionsystemesfaf培养基培养下hi5细胞表达的pepr1体外纯化结果图,a图为利用凝胶过滤层析方法纯化pepr1蛋白,pepr1蛋白的凝胶层析结果,横纵坐标分别代表洗脱体积和紫外吸收值(λ=280),pepr1正确洗脱位置如箭头所示。b图为图a对应洗脱位置处收集的样品进行sds-page和考马斯亮蓝染色检测。
图6为不同培养基(sf900ⅱ)下pepr1蛋白的表达情况。a图为利用凝胶过滤层析方法纯化pepr1蛋白,pepr1蛋白的凝胶层析结果,横纵坐标分别代表洗脱体积和紫外吸收值(λ=280),pepr1正确洗脱位置如箭头所示。b图为图a对应洗脱位置处收集的样品进行sds-page和考马斯亮蓝染色检测。与图5相比,pepr1蛋白正确洗脱位置处uv吸收值明显降低为expressionsystemesfaf培养基的0.75%。
图7为pepr1小量表达检测(+dtt和-dtt对比)。hi5细胞表达的pepr1的小量检测实验图。细胞培养液经镍柱纯化后洗脱液进行sds-page和考马斯亮蓝染色,两条泳道分别是对pepr1蛋白的还原性(+dtt)和非还原性(-dtt)检测结果。
图8为pepr1蛋白的westernblot检测结果。
具体实施方式
以下实施例用于进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。
若无特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
实施例1pepr1基因外显子的获得
考虑到pepr1胞外编码区不存在内含子区域,本发明从拟南芥基因组中直接扩增出的pepr1的胞外结构域。pcr扩增实验选用高保真的pcr酶,以防在扩增过程中发生碱基突变,所用引物序列如下。
pepr1_29_bamhi_5:cgcggatccttaaactcagatgggctaac
(seqidno.4)
pepr1_767_6his_sali_3t:aggaagagtggccttagcacccatcatcatcatcatcattaagtcgacgtc(seqidno.5)
pcr扩增反应体系:5μl10×pfu缓冲液,2.5μldntp(2.5mm),0.1μm的上下游引物各0.5μl,pfu聚合酶1μl,模板0.5μl,加双蒸水至50μl。pcr扩增反应条件为95℃,5min;共30个循环:94℃30s,53℃30s,72℃600bp/min计算所需延伸时间;72℃10min。pepr1基因的pcr扩增产物凝胶电泳图见图2。按照试剂盒说明书操作回收pcr扩增产物。
对pepr1基因序列进行结构域分析,采用信号肽预测网站(signalp)和疏水跨膜区预测网站(tmhmm)预测出pepr1的疏水跨膜区和信号肽区域。在疏水跨膜区前的这一段区域即为胞外区。考虑到pepr1蛋白属于分泌蛋白,其本身的n端信号肽会带领着合成好的肽链在内膜系统中进行后期修饰和加工,并且引领加工好的蛋白分泌到细胞膜外,当信号肽的功能完成后,信号肽会在后期被剪切掉,成为成熟的分泌蛋白。通过软件预测后初步确定pepr1跨膜区为770-792aa。跨膜区前端边界处可以前后3-4个氨基酸多做几个片段长度,以防预测位置与实际切割位置有误差造成蛋白错误折叠。最终确定的pepr1胞外区长度是1-767aa,其氨基酸序列如seqidno.1所示,其核苷酸序列如seqidno.2所示。
实施例2外源表达载体pfastbactmhem的构建
信号肽在蛋白表达加工过程中发挥重要作用,将pepr1这类植物蛋白在昆虫细胞中进行外源表达过程中,由于无法得知植物蛋白自身的信号肽转入到昆虫细胞中是否依然可以正常行使自身的功能,故本实施例将昆虫细胞表达系统中的杆状病毒表达载体pfastbactm1(图3)进行了改造,新改造好的载体为pfastbactm-hem(图4a和图4b)。即在pfastbactm1载体上与目的基因n端连接的前端部分上加入一段杆状病毒来源的抑血细胞聚集素(hemolinpeptide)信号肽,其核苷酸序列如seqidno.3所示。
本发明意外发现采用hem信号肽比采用pepr1自身的n端信号肽效果要更好,可以被昆虫体系识别、剪切并引导穿越内质网膜的序列作为分泌信号肽。同时本发明在目的基因的c末端加上便于纯化的his标签,采用在3’-5’端引物上加上6个组氨酸密码子序列和终止密码子序列,使阅读框一直向下读取直至到读取完his标签的密码子,然后读取完终止密码子。翻译结束。
选用pfastbactmhem这一本发明新改造的载体作为表达载体,后期与目的基因连接时,需要将连入的目的基因片段(pepr1外显子基因)前的自身信号肽去掉,因此连入的目的片段是pepr1-29-767,去掉了n端的28个氨基酸。
实施例3pepr1基因外显子与外源表达载体pfastbactmhem连接与重组bacmid的制备
限制性内切酶的选择是建立在目的基因序列中不能够出现所挑选的位于基因两侧的这两种限制性内切酶的酶切序列,否则在进行后期的酶切实验过程中,所要连入载体的基因序列会被限制性内切酶所切割,从而无法得到成功的重组质粒。基于上述原则,本实施例在筛选酶切位点时,选择了两对限制性内切酶bamh1和xho1,bgl2和sal1。bamh1和bgl2互为同尾酶,xho1和sal1互为同尾酶。
这两组同尾酶对实施例1获得的目的基因进行酶切,每一组同尾酶中各选取一个,两两搭配进行使用。共有四种组合方式:bamh1和xho1,bamh1和sal1,bgl2和xho1,bgl2和sal1。
同时本发明发现在实施例1获得的pepr1基因中没有bamh1和sal1这两种限制性内切酶对应的酶切位点。因此选择bamh1和sal1这个组合进行酶切。前述四种组合的合理运用可以最大限度地规避目的基因中出现酶切位点序列,减少后期采用点突变来消除酶切位点的工作量。
外源载体为实施例2改造得到的载体pfastbactmhem采用bamh1和xho1这两种限制性内切酶进行处理后备用。而目的基因两端的酶切位点选择bamh1和sal1,这样目的片段才能顺利连入已切割的载体。后期采用bamh1和sal1这两种限制性内切酶对pepr1进行切割时,pepr1基因不会被这两种酶所切断。然后根据密码子的容错性原理将碱基进行点突变,这样既消除该酶切位点,同时也并不会造成氨基酸的突变。选择了酶切位点后,在(酶切位点的)5’和3’端再分别加上合适的保护碱基即可。选择上述酶切位点后,对外源载体和pepr1基因的酶切方法均采用本领域常规技术,获得pepr1基因与载体pfastbactmhem的连接产物。
本发明的pepr1蛋白是选用pfastbactmhem载体加上pepr1-29-767基因片段,能够获得正确折叠表达的pepr1胞外区蛋白,并提高pepr1的表达量,表达量达到2.24mg/l。
从-80℃冰箱中取出制备好或购买的感受态细胞bl21(de3)或dh5α,将准备转化的连接产物加入到100ul或25ul感受态细胞中(根据感受态细胞的浓度和效率而定),在冰上放置15分钟,然后在42℃水浴锅中温浴90秒,立马取出放于冰上5分钟,加入600μllb液体培养基,37℃180转/分钟振荡复苏培养1个小时,吸取培养液均匀涂在具有氨苄抗生素的lb平板上,37℃温箱过夜培养。挑取一定数量的单克隆菌落于装有1ml相应抗性培养基的ep管中,37℃220转/分钟振荡复苏培养10-12个小时,取1ul菌液做模版进行pcr鉴定,或者提取质粒进行酶切鉴定,经鉴定为阳性的单克隆送测序公司测序,测序正确的克隆,保存质粒于-20℃冰箱待用。
将上步测序正确的质粒转化到感受态细胞dh10bac,到复苏培养时,为保证重组的充分,需要复苏5-6个小时,之后取30ul培养基,用灭菌后的玻璃棒均匀涂在三种抗生素的(卡那霉素,庆大霉素,四环素)平板上,放于37℃温箱中避光培养40-48个小时。40-48个小时后,可清晰看出蓝色或者白色的斑点,挑取白色斑点于三抗培养基中,37℃培养后,取1ul菌液做模版,进行pcr鉴定,引物一端来自基因扩增的引物(seqidno.4或5),另外一端为bacmid上固有的m13序列引物,片段大小正确的阳性克隆是重组的bacmid。鉴定阳性后,根据试剂盒说明书提取病毒粘粒bacmid,提取后取1-2μl重组bacmid进行琼脂糖凝胶电泳,确认提取的重组bacmid的含量是否足够,重组杆状病毒粘粒放于4℃保存备用。
实施例4pepr1蛋白的表达
1、病毒的包被
接下来进行病毒的制作,病毒的制作是采用昆虫细胞来制作的,因此首先准备好位于适宜生长期和适宜浓度的昆虫细胞。在本实验中共采用了两种昆虫细胞,分别是sf21和hi5昆虫细胞(见表1)。
表1细胞株列表
sf21细胞可以用于病毒的制作和培养表达;hi5昆虫细胞只用于培养表达。昆虫细胞采用悬浮培养方式,为对数式生长。病毒的制作过程叫做转染,所有操作均在细胞间超净台内进行,具体步骤如下:
(1)配制溶液a:取一个新的1.5ml灭菌离心管,在管中加入5-7μl实施例3制得的杆状病毒粘粒(pepr1重组bacmid),再加入100μl未加抗生素(链霉素和青霉素)的sf900ii培养基(购自gibco公司,本实施例细胞培养基均指代sf900ii培养基),轻轻吹吸混匀。
(2)配制溶液b:取一个新的1.5ml灭菌离心管,在管中加入5-7μl转染试剂(cellfectin,购自invitrogen),再加入100μl未加抗生素的sf900ii培养基,轻轻吹吸混匀。如果转染bacmid的个数较多,可以配置一个mix溶液备用。mix溶液成分包括转染试剂和无抗的sf900ii培养基,溶液b的mix溶液,将溶液b的各成分按照比例放大;例如在单次实验操作中实验者需要做5种不同杆状病毒粘粒的转染,可以准备5份剂量溶液b的mix溶液,目的在于保证每份带有杆状病毒粘粒的溶液a都可以与完成相同的溶液b混合,减少人工操作的误差。
(3)将溶液b加入溶液a中,轻轻吹吸混匀,命名为溶液c。将溶液c放入28℃培养箱静置30分钟。
(4)选取状态良好处于对数生长期的sf21细胞,细胞浓度1.5~2.5×106个细胞/毫升,铺于细胞6孔板,每个孔细胞总数控制为1~1.5×106个,并最后将细胞孔内的培养基体积补齐至2ml体积,前后或者左右轻晃培养板,使细胞均匀分布于孔内。将培养板放入28℃培养箱静置10分钟左右,使细胞充分贴壁。
(5)待细胞完全贴壁后,吸去原有的培养基,每个孔加入1-1.5ml无抗培养基,轻轻晃动六孔板,充分润洗细胞。此步骤的目的是除去原有培养基中的抗生素,因为抗生素会影响转染效率。吸去六孔板中的润洗培养基,再加入1ml无抗培养基待用。
(6)将溶液c轻轻吹吸混匀后加到六孔板的细胞里,轻晃六孔板使其均匀覆盖。
(7)将六孔板置于28℃培养箱中,静置5小时。
(8)换液。将含有转染试剂和粘粒的培养基吸走,每个孔加入2-3ml含有青霉素和链霉素抗性的sf900ii培养基。继续在28℃培养箱中静置培养3-4天。
(9)观察转染现象,并收取第一代病毒,吸取培养液于15ml离心管,2000转/分离心5分钟,上清即为重组pepr1杆状病毒,命名为p1,避光保存于4℃。为观察转染现象是否明显,可做对照组实验,有利于比较。病毒转染后培养时间过长会细胞裂解,内容物释放影响病毒状态,静置培养的时间最好不要超过5天。p1病毒,量少且滴度低,无法用于大量表达目的蛋白,因此需要扩增p2(第二代)和p3(第二代)病毒。通常情况下,病毒只扩增到第三代,因为继续扩增的病毒表达外源蛋白的能力会有所下降。p2病毒用于目的蛋白小量检测,确定表达后,扩增p3病毒,用于目的蛋白的大量纯化,评价蛋白行为等等。
本发明采用的bac-to-bac杆状病毒表达系统的过程示意图见图1。
2、蛋白的表达
2.1昆虫细胞小量检测目的蛋白的表达
在大量培养纯化蛋白之前,先需要采用小量检测一下蛋白是否表达以及蛋白表达量的多少;一般用p2或p3病毒进行蛋白表达测试,测试的细胞为悬浮培养,细胞的种类sf21和hi5细胞均可以选择,hi5细胞更适用于分泌蛋白的表达。测试的方法及实验条件如下(由于本研究中最后比较的结果是hi5细胞表达的效果更好,因此这里以hi5细胞的表达为例):
(1)细胞密度:hi5细胞对于密度要求更为严格,最好在1.6-2.0×106个/ml之间。确保细胞状态良好并处于对数生长期。
(2)待细胞长到合适的密度后,每个玻璃培养瓶分装25-30ml细胞,每瓶细胞中加入p2或p3病毒。若没有测过病毒滴度,则加入的病毒量需做梯度测试,不同的蛋白或者不同批次的病毒情况可能会有所不同。起始可以按照病毒:细胞体积为1:50加入。
(3)培养的温度和培养时间:hi5细胞在病毒未侵染前在28℃悬浮培养,120转/分;当加入病毒侵染后将侵染后在细胞放置在22℃悬浮培养,120转/分;侵染时间48小时。48小时后采用4000转/分,离心10分钟分离上清和细胞。若表达分泌蛋白,是被分泌到培养基中的,则离心后弃细胞沉淀,取上清;反则取沉淀。分泌表达蛋白,上清直接倒入200μl亲和柱beads;再加入2次2ml的washbuffer,最后用300ulelutionbuffer流过亲和柱,流过的elutionbuffer留存下来,加上loadingbuffer上样,用sds-page检测。
对于pepr1这种分泌蛋白,在sds-page检测上可以用一个小策略,能够简单快速地了解正确构象的分泌蛋白的真实表达量是怎么样的。在常规的sds-page实验中采用的蛋白样品loadingbuffer组分里是含有dtt的,为了将蛋白质中的二硫键打开,使实验者更容易观察蛋白样品的分子量大小。但是对于pepr1这种分泌蛋白来说,外源表达的过程中经常会伴有蛋白聚集体的出现,采用不含有dtt的loadingbuffer可以在不使用凝胶过滤层析实验之前就可以分辩外源获得的pepr1蛋白是构象正确的蛋白还是构象异常的蛋白聚集体。上样时使用的loadingbuffer的配方里去掉dtt(二硫代苏糖醇),由于二硫代苏糖醇的作用是打开蛋白质中形成的二硫键。分泌类蛋白存在着许多二硫键,二硫键容易发生错误配对从而导致蛋白构象变化形成聚集体,当加入dtt后再通过sds-page检测蛋白会错误将一些二硫键错配形成的蛋白也认为是正确构象的蛋白,因此去掉dtt后的sds-page上分子量大小正确的位置上蛋白条带的深浅才是正确构象蛋白的表达量,通过对+dtt和-dtt的比对,可以了解正确构象蛋白在蛋白总量中的比例。本实施例通过比较加dtt和未加ddt进行sds-page结果的方法来定性地判断在page胶上哪个条带是构象正确的pepr1蛋白,然后根据该条带的宽度和深浅差异大概估量出正确构象的pepr1蛋白的表达量,因为洗脱下来的蛋白会包含构象正确的pepr1蛋白,构象异常的聚集体及一些杂蛋白,有很多条带在胶上,先要定性哪条条带是构象正确的蛋白,然后再根据宽度和深浅来大概定量,实际表达量的数值需要参考-dtt的蛋白条带的深浅和宽度。见图7
通过凝胶过滤层析实验结果中蛋白的洗脱位置和sds-page结果中的蛋白分子量大小判断所表达蛋白的构象是正确的,如图5,pepr1洗脱位置在凝胶层析结果上已标明,出峰位置在62-69ml之间,sds-page上蛋白分子量是114kda,与pepr1的实际分子量相符。由于pepr1蛋白是带有his标签的,利用his标签的抗体进行杂交实验,在114kda的位置检测到蛋白,说明本发明获得蛋白是带有his标签的pepr1蛋白。见图8。
根据检测结果确定蛋白是否表达以及最优化的细胞系(在sf21细胞和hi5细胞两种细胞系中选择表达量更多,杂蛋白更少的hi5细胞系。hi5细胞系相比较于sf21细胞系,hi5细胞系在表达外源蛋白时更适于用于分泌型蛋白的表达,杂蛋白量更少,构象正确的蛋白表达量更高,在本实施例中直接在两者中选择hi5细胞系来用于表达pepr1蛋白。
2.2蛋白的大量培养和纯化
根据小量检测目的蛋白最后的优化情况,将表达体系扩至500~600ml,用于检测目的蛋白的蛋白行为和蛋白量,最后扩大培养至2~3l再进行大量培养,以得到足够量的蛋白供后期实验所用。大量培养时和小量检测时所采用的培养基为gibco公司的sf900ⅱ无血清培养基(1×)1000ml,当使用时先加入5ml青霉素链霉素混合液(gibco公司),以防止在使用培养基传代时造成细胞污染。在培养细胞时发现可以不向细胞培养基内添加胎牛血清(fbs),并不会影响昆虫细胞的正常生长和外源表达功能。
本发明中提供了一种新型的昆虫细胞培养方法能够显著提高正确构象的pepr1蛋白的表达量。具体方法如下:hi5细胞的侵染浓度为1.6-2.0×106个/ml之间,在病毒侵染的前一天下午至晚上这一时间段,采用义翘神州公司购买的expressionsystemesfaf培养基代替sf900ⅱ细胞培养基将hi5细胞(浓度大概在2.5-3.2×106个/ml)进行扩大培养,稀释至细胞浓度大概在0.3×106个/ml,过夜培养;第二天计数当生长到1.6-2.0×106个/ml时,进行侵染。后续步骤中培养时间、培养温度和转速与上文中hi5细胞在sf900ⅱ细胞培养基中的培养步骤完全一致。采用上述方法培养细胞后得到的蛋白表达量与采用gibco公司的sf900ⅱ无血清培养基来培养细胞后得到的蛋白表达量差异可以参考图5和图6。与图5相比,图6中pepr1蛋白正确洗脱位置处uv吸收值明显降低为expressionsystemesfaf培养基的0.75%。说明采用expressionsystemesfaf培养基进行昆虫细胞扩大培养后,再转入含pepr1的外源表达载体,能够显著提高正确构象的pepr1蛋白的表达量。
由于本研究中蛋白pepr1为分泌表达的蛋白。在目的基因的c端,加了6×his纯化标签,故用ni-ntahis标签结合介质纯化目的蛋白。分泌蛋白的提取过程如下(提取过程在4℃冷室进行):
(1)用大容量离心机4000转/分,离心10分钟收获上清后,将上清加到再生好的ni-nta亲和柱中,使上清依靠重力作用缓慢通过亲和介质。为保证结合效率,每100ml培养基用1根ni-nta亲和柱,且上样两遍。每根ni-nta亲和柱柱料2~3ml。
(2)用10ml含有15mm咪唑的ni-nta漂洗缓冲液润洗亲和介质。
(3)重复步骤(2)两次,尽可能地洗去杂蛋白。
(4)用5ml的ni-nta洗脱缓冲液洗脱介质上结合的蛋白两次。蛋白洗脱液留作进一步纯化并取样,标记为elution(e)。
随后采用凝胶过滤层析的方法来进一步纯化的目的蛋白,采用的是ge公司的actapurifer自动上样洗脱系统,系统上自带上样loop的上样体积为2ml,首先需要蛋白浓缩管将elution浓缩到上样体积2ml以内。采用分离蛋白量适宜的(10kd,30kd和50kd)种类,例如目的蛋白为100kd,最后采用10kd,30kd比较适合;采用分子量较小的浓缩管会影响浓缩的速度,过大的话会导致部分目的蛋白的损失,采用低温4℃离心机,3200-4000转/min进行浓缩。
将样品加入到上样loop里后,设置好程序就可以自动进行上样和洗脱过程,凝胶过滤层析是根据蛋白质分子的分子量的差异进行分离的,不同的层析柱体积和不同规格柱料会产生不同的分离效果,在实验室常用的两种规格(hiloadtm16/600superdextm200pg和superdextm20010/300gl)的层析柱中,superdextm20010/300gl适于分离20~600kd的蛋白;hiload200适用于大量分离和制备分子量约为20~600kd的蛋白,在分离60-120kd大小的蛋白质时,hiload200较superdextm20010/300gl效果更好;经过实验结果显示,pepr1胞外区蛋白更适于采用hiload200来分离纯化。
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
序列表
<110>中国农业科学院蜜蜂研究所
<120>一种外源表达pepr1蛋白的方法
<160>5
<170>siposequencelisting1.0
<210>1
<211>739
<212>prt
<213>人工序列(artificialsequence)
<400>1
leuasnseraspglyleuthrleuleuserleuleulyshisleuasp
151015
argvalproproglnvalthrserthrtrplysileasnalaserglu
202530
alathrprocysasntrppheglyilethrcysaspaspserlysasn
354045
valalaserleuasnphethrargserargvalserglyglnleugly
505560
progluileglygluleulysserleuglnileleuaspleuserthr
65707580
asnasnpheserglythrileproserthrleuglyasncysthrlys
859095
leualathrleuaspleusergluasnglypheserasplysilepro
100105110
aspthrleuaspserleulysargleugluvalleutyrleutyrile
115120125
asnpheleuthrglygluleuprogluserleupheargileprolys
130135140
leuglnvalleutyrleuasptyrasnasnleuthrglyproilepro
145150155160
glnserileglyaspalalysgluleuvalgluleusermettyrala
165170175
asnglnpheserglyasnileprogluserileglyasnserserser
180185190
leuglnileleutyrleuhisargasnlysleuvalglyserleupro
195200205
gluserleuasnleuleuglyasnleuthrthrleuphevalglyasn
210215220
asnserleuglnglyprovalargpheglyserproasncyslysasn
225230235240
leuleuthrleuaspleusertyrasngluphegluglyglyvalpro
245250255
proalaleuglyasncysserserleuaspalaleuvalilevalser
260265270
glyasnleuserglythrileproserserleuglymetleulysasn
275280285
leuthrileleuasnleusergluasnargleuserglyserilepro
290295300
alagluleuglyasncysserserleuasnleuleulysleuasnasp
305310315320
asnglnleuvalglyglyileproseralaleuglylysleuarglys
325330335
leugluserleugluleuphegluasnargpheserglygluilepro
340345350
ilegluiletrplysserglnserleuthrglnleuleuvaltyrgln
355360365
asnasnleuthrglygluleuprovalglumetthrglumetlyslys
370375380
leulysilealathrleupheasnasnserphetyrglyalailepro
385390395400
proglyleuglyvalasnserserleuglugluvalasppheilegly
405410415
asnlysleuthrglygluileproproasnleucyshisglyarglys
420425430
leuargileleuasnleuglyserasnleuleuhisglythrilepro
435440445
alaserileglyhiscyslysthrileargargpheileleuargglu
450455460
asnasnleuserglyleuleuproglupheserglnasphisserleu
465470475480
serpheleuasppheasnserasnasnphegluglyproileprogly
485490495
serleuglysercyslysasnleuserserileasnleuserargasn
500505510
argphethrglyglnileproproglnleuglyasnleuglnasnleu
515520525
glytyrmetasnleuserargasnleuleugluglyserleuproala
530535540
glnleuserasncysvalserleugluargpheaspvalglypheasn
545550555560
serleuasnglyservalproserasnpheserasntrplysglyleu
565570575
thrthrleuvalleusergluasnargpheserglyglyileprogln
580585590
pheleuprogluleulyslysleuserthrleuglnilealaargasn
595600605
alapheglyglygluileproserserileglyleuilegluaspleu
610615620
iletyraspleuaspleuserglyasnglyleuthrglygluilepro
625630635640
alalysleuglyaspleuilelysleuthrargleuasnileserasn
645650655
asnasnleuthrglyserleuservalleulysglyleuthrserleu
660665670
leuhisvalaspvalserasnasnglnphethrglyproileproasp
675680685
asnleugluglyglnleuleusergluproserserpheserglyasn
690695700
proasnleucysileprohisserpheseralaserasnasnserarg
705710715720
seralaleulystyrcyslysaspglnserlysserarglyssergly
725730735
leuserthr
<210>2
<211>2301
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgaagaatcttggggggttgttcaaaattcttctgcttttcttctgtctctttctatcg60
acccacataatttccgtttcttgtttaaactcagatgggctaactctactctctcttctg120
aagcatttggatagagtaccaccacaagttacttcgacatggaaaataaacgcatctgaa180
gcaactccatgtaactggttcggtatcacttgtgacgattctaagaatgttgcgtctctc240
aacttcactcgttctagggtttcaggtcaattgggtccggaaattggggagctcaaaagc300
ttgcagattttggatctgagtactaacaatttctccgggactataccttccactttagga360
aactgtaccaaactcgctactctagatttgtctgaaaatggattctctgataagatccca420
gatactctcgatagcttgaagaggttggaggtgctttatctttacataaacttcctcact480
ggtgagttacctgaatccttgtttcgaattccgaagctgcaggttttatatcttgactat540
aacaatctcaccggtccgattcctcaaagtattggtgatgctaaggagcttgtggagctg600
agtatgtatgcgaatcagttctctggtaacatccctgagtcgattgggaatagcagtagt660
ctgcagattctttatttgcacaggaacaagttagttggttcattacctgaaagtctcaat720
cttttggggaatctcactactctgtttgttggtaacaacagtctacaagggccggttcgt780
ttcggatcacctaattgcaagaatttgttgactttagatttgtcatacaatgaattcgaa840
ggcggtgttccacctgcattgggaaattgcagtagccttgacgctttagtcattgtgagt900
ggtaacttgtcaggtacaatcccttcctcattgggtatgttgaagaatctcacaattctt960
aacctttccgagaatcgtctctctgggagtatccccgcagagctcgggaactgcagtagc1020
ttgaacttgttgaagctgaacgataaccagcttgtaggcggaataccgagtgcattaggt1080
aagctgaggaagctagaaagtctggagcttttcgaaaaccggttttcgggtgagattcct1140
attgagatatggaagagtcagagtcttacgcagttgctagtttatcaaaacaatctcact1200
ggtgaactacctgtggaaatgactgagatgaagaagctaaagatcgctacgctgttcaac1260
aacagcttttatggagcgataccaccgggtttaggtgtgaacagcagcttagaagaggtt1320
gactttattggtaacaaacttacaggagagataccgccaaatctatgccatggaaggaag1380
ttgagaatactcaacttgggttctaatctgcttcatggtacaataccagcttctattggt1440
cactgtaagaccatcaggagattcatccttagagaaaataacctttcaggtcttcttcct1500
gagttttctcaggatcatagtctttcttttcttgatttcaatagcaacaacttcgaagga1560
ccaatcccgggcagcctcggaagctgtaagaatctctcgagtattaacctatctcgaaac1620
agattcacggggcagatacctccacaacttgggaatctacaaaaccttggttacatgaat1680
ctttctcgtaatcttcttgaagggtctctaccagctcagctatctaactgtgtgagttta1740
gagcgttttgatgttggcttcaactcattaaacggttcagttccttcaaactttagtaac1800
tggaaaggcttgacgactttagttctcagcgagaaccggttttcaggaggtattccacag1860
ttcttgcctgagcttaagaagctgtcaactctgcagattgctagaaatgcttttggtggt1920
gagattccttcgtcgattgggttgatagaggatctgatctatgacttggaccttagtgga1980
aacggattgacaggtgaaattccagccaagttgggagatctcatcaagttaacaagactc2040
aacatatctaacaacaatttgacaggatctttatcggttctcaaaggtcttacctcattg2100
ctacatgttgatgtctccaacaatcagttcacaggtccaataccagataacttggagggt2160
cagttgttatctgagccgtcgtcgttttcaggaaatccaaacctctgcattccacattcc2220
ttctctgctagcaacaatagccgcagcgcgttaaagtactgtaaagatcaatctaaaagc2280
aggaagagtggccttagcacc2301
<210>3
<211>60
<212>dna
<213>人工序列(artificialsequence)
<400>3
atggcgttcaagagtatagcagttttaagcgcctgcataattgtgggttcagcgcttccg60
<210>4
<211>29
<212>dna
<213>人工序列(artificialsequence)
<400>4
cgcggatccttaaactcagatgggctaac29
<210>5
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>5
aggaagagtggccttagcacccatcatcatcatcatcattaagtcgacgtc51
- 还没有人留言评论。精彩留言会获得点赞!