五氟磺草胺原药相关杂质bis-CHYMP的制备方法与流程
本发明涉及一种五氟磺草胺原药相关杂质bis-chymp的制备方法。
背景技术:
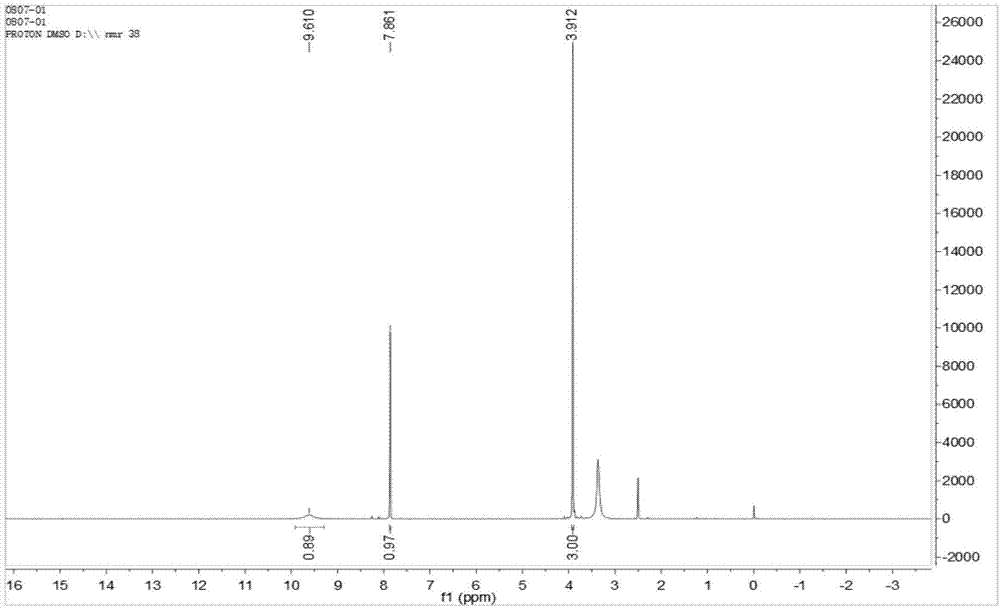
:bis-chymp,全称为2-chloro-4-[2-(2-chloro-5-methoxy-4-pyrimidinyl)hydrazi--no]-5-methoxypyrimidine,即2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶(cas:1956386-49-4),是五氟磺草胺原药中指定检测的相关杂质,其化学架构见结构式1。结构式1:五氟磺草胺(penoxsulam)是由美国陶氏益农公司(dowagrosciences)开发的三唑并嘧啶磺酰胺类传导型除草剂,使用后经茎叶、幼芽及根系吸收,通过木质部和韧皮部传导至分生组织,抑制乙酰乳酸合成酶的作用,从而造成杂草植株生长停止、黄化并死亡,以此作用来除去水稻田中的杂草。该产品于2004年在美国epa正式注册登记,在2008年在我国登记,国内厂家将其制成悬浮剂进行销售。在农药出口登记和国际贸易中,各国政府对农药原药和制剂中相关杂质的种类和浓度尤其关注,相关杂质为必检项目,为此fao规定了五氟磺草胺的相关杂质bis-chymp的限量,目前已有文献通过高效液相色谱法和液质联用的方法对五氟磺草胺原药进行定性定量的研究,在检测过程中需要用bis-chymp做为标准品,现在国内对bis-chymp的研究较少,尤其是其合成过程,至今未有报道。因此,急需一种反应条件温和,耗能较少,经济可行,产率较高,纯度较高的五氟磺草胺原药相关杂质bis-chymp的制备方法。技术实现要素:为了解决五氟磺草胺中相关杂质bis-chymp制备工艺复杂,产率较低的问题,本发明的目的是提供一种五氟磺草胺原药相关杂质bis-chymp的制备方法,具有反应条件温和,耗能较少,经济可行,产率较高,纯度较高的优点。本发明提供了如下的技术方案:五氟磺草胺原药相关杂质bis-chymp的制备方法,包括如下步骤:s1、将2,4-二氯-5-甲氧基嘧啶溶解于甲醇中,然后加入三乙胺进行搅拌,溶解完全后再加入水合肼,进行回流反应,tlc监测反应完成后冷却至室温,萃取、洗涤和干燥,柱层析分离纯化得到中间体2-氯-5-甲氧基-4-肼基嘧啶;s2、将2,4-二氯-5-甲氧基嘧啶溶解于甲醇中配制2,4-二氯-5-甲氧基嘧啶的甲醇溶液,将所述s1步骤制备的2-氯-5-甲氧基-4-肼基嘧啶溶解在甲醇中,加入三乙胺进行搅拌,溶解完全后,缓慢滴加配制好的2,4-二氯-5-甲氧基嘧啶的甲醇溶液,进行回流反应,lcms检测反应结束后,降温冷却,得到反应后的混合液;s3、将所述s2步骤得到的混合液经过萃取、洗涤和浓缩工序的后处理,再经过制备型高效液相进行分离纯化,得到棕色固体2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶;合成路线为:本发明的技术方案是是采用2,4-二氯-5-甲氧基嘧啶为起始原料,以甲醇为溶剂,与三乙胺和水合肼进行反应得到中间体2-氯-5-甲氧基-4-肼基嘧啶,然后再以甲醇为溶剂,2-氯-5-甲氧基-4-肼基嘧啶、2,4-二氯-5-甲氧基嘧啶和三乙胺进行反应,终产物经过萃取、洗涤和浓缩的后处理工序,最后经过制备型高效液相色谱纯化,最终制得产物2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶,即bis-chymp,收率达到40%,本发明两步反应均用甲醇做溶剂,甲醇沸点65℃左右,反应条件温和,耗能较少,经济可行,且具有产率较高的优点。优选的,所述s1步骤中2,4-二氯-5-甲氧基嘧啶:水合肼:三乙胺的摩尔比为1:1:1.05:3。优选的,所述s1步骤中回流反应时间为12h。优选的,所述s1步骤中tlc监测的柱层析展开剂采用二氯甲烷-甲醇-三乙胺(10:1:0.05)。优选的,所述s2步骤中2-氯-5-甲氧基-4-肼基嘧啶:2,4-二氯-5-甲氧基嘧啶:三乙胺的摩尔比为1:1:3。优选的,所述s2步骤中回流反应时间为24h。优选的,所述s3步骤中制备型高效液相的色谱条件为:采用制备柱为xbridgeprepc185μmobd30×150mmcolumn;进样量3000μl;检测波长282nm;流速为35ml/min;收集组分时间为13.7-15.4min;以乙腈为流动相a,以0.1%甲酸水溶液为流动相b;梯度洗脱程序为:0-5min,流动相a和流动相b的体积百分数分别为10%和90%,5-20min,5-20min流动相a的体积百分数由10%至90%。梯度洗脱程序t/min乙腈(a)0.1%甲酸水(b)0109051090209010本发明的有益效果是:本发明的技术方案是是采用2,4-二氯-5-甲氧基嘧啶为起始原料,以甲醇为溶剂,与三乙胺和水合肼进行反应得到中间体2-氯-5-甲氧基-4-肼基嘧啶,然后再以甲醇为溶剂,2-氯-5-甲氧基-4-肼基嘧啶、2,4-二氯-5-甲氧基嘧啶和三乙胺进行反应,终产物经过萃取、洗涤和浓缩的后处理工序,最后经过制备型高效液相色谱纯化,最终制得产物2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶,即bis-chymp,产率达到40%,本发明两步反应均用甲醇做溶剂,甲醇沸点65℃左右,反应条件温和,耗能较少,经济可行,且具有制备的杂质纯度较高,产率较高的优点。附图说明附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:图1为bis-chymp的1hnmr图谱。图2为bis-chymp的13cnmr图谱。图3为bis-chymp的mass图谱。图4为bis-chymp的高效液相色谱图。具体实施方式实施例1五氟磺草胺原药相关杂质bis-chymp的制备方法,包括如下步骤:s1、取一500ml三口瓶,加入0.01mol的2,4-二氯-5-甲氧基嘧啶(1.79g),加入200ml无水甲醇,然后加入0.03mol的三乙胺(3.03g/4.15ml),进行搅拌溶解,溶解完全后,用恒压滴液漏斗缓慢滴加0.01mol水合肼(27%,1.81g)至反应液中,约30分钟之内滴加完毕,升温至回流状态,进行回流反应,反应时间为12h,tlc进行监测,tlc监测的柱层析展开剂采用二氯甲烷-甲醇-三乙胺(10:1:0.05),tlc监测反应完成后冷却至室温,萃取、洗涤和干燥,柱层析分离纯化得到中间体2-氯-5-甲氧基-4-肼基嘧啶(1.12g),收率为65%;s2、将0.01mol2,4-二氯-5-甲氧基嘧啶(1.79g)溶解于200ml甲醇中配制2,4-二氯-5-甲氧基嘧啶的甲醇溶液,将所述s1步骤制备的0.01mol2-氯-5-甲氧基-4-肼基嘧啶(1.74g)溶解在400ml甲醇中,加入0.03mol三乙胺(3.03g/4.15ml)进行搅拌,溶解完全后,用恒压滴液漏斗缓慢滴加配制好的2,4-二氯-5-甲氧基嘧啶的甲醇溶液,约30分钟滴加完毕,升温至回流状态,进行回流反应,反应时间为24h,lcms检测反应结束后,降温冷却,得到反应后的混合液;s3、将所述s2步骤得到的混合液经过萃取、洗涤和浓缩工序的后处理,再经过制备型高效液相进行分离纯化,制备型高效液相的色谱条件为:采用制备柱为xbridgeprepc185μmobd30×150mmcolumn;进样量3000μl;检测波长282nm;流速为35ml/min;收集组分时间为13.7-15.4min;以乙腈为流动相a,以0.1%甲酸水溶液为流动相b;梯度洗脱程序为:0-5min,流动相a和流动相b的体积百分数分别为10%和90%,5-20min,5-20min流动相a的体积百分数由10%至90%,梯度洗脱程序如下表1,表1梯度洗脱程序t/min乙腈(a)0.1%甲酸水(b)0109051090209010分离纯化后得到棕色固体2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶(1.26g),收率40%,收率较高。实施例2(1)用核磁氢谱、核磁碳谱和质谱对产物2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶进行结构确证。用核磁氢谱(brukerav-400型核磁共振仪,dmso-d6)测定产物的核磁共振氢谱,图谱如图1所示,1h-nmr:δppm3.912(och3,s,6h),7.861(pyrim-h,s,2h),9.610(nh,s,2h);用核磁碳谱(brukerav-400型核磁共振仪,dmso-d6)测定产物的核磁共振碳谱,图谱如图2所示,13c-nmr:δppm56.384,135.102,139.085,150.190,155.005;用质谱(waterszq2000,溶剂甲醇,esi(+)70v)317.0[m+h]+,339.0[m+na]+测定产物的质谱,图谱如图3所示,从质谱esi(+)结果[m+h]+的m/z为317.0,可以推测样品分子量为316,与c10h10cl2n6o2理论分子量相符,符合奇氮原子奇分子量的氮规则,最终可确证产物为2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶。(2)采用高效液相色谱测定产物2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶的纯度,高效液相色谱图如图4所示,可知2-氯-4-[2-(2-氯-5-甲氧基-4-嘧啶碱基)肼基]-5-甲氧基嘧啶的归一化含量为99.9%,纯度较高。以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1