氮杂糖及其中间体的合成的制作方法
本发明涉及用于制备可用于合成半乳糖苷酶抑制剂米加司他(migalastat)的中间体的方法。
背景技术:
式(i)的(2r,3s,4r,5s)-2-(羟甲基)-3,4,5-三羟基哌啶,
也被称为1,5-二脱氧-1,5-亚氨基-d-半乳糖醇、1-脱氧半乳糖野尻霉素(1-deoxygalactonojirimycin,dgj)或米加司他,是α-半乳糖苷酶和β-半乳糖苷酶的强效抑制剂,所述α-半乳糖苷酶和β-半乳糖苷酶是复杂碳水化合物代谢的重要酶。已经在治疗各种疾病方面研究了所述酶的抑制剂,所述疾病包括糖尿病(u.s.4,634,765)、癌症(u.s.5,250,545)、疱疹(u.s.4,957,926)、hiv或法布里病(fabrydisease)(fan,j.q.等人《自然·医学(nat.med.)》1999,5,112)。
米加司他是亚氨基糖或氮杂糖的代表,亚氨基糖或氮杂糖是包含一个环内氮原子的多羟基化杂环家族。这些化合物的首次合成报道于1960年代初(paulsen,h.,《应用化学国际英语版(angew.chem.int.ed.engl.)》1962,1,454;jones,j.k.n.等人,《加拿大化学杂志(can.j.chem.)》1963,41,636;hanessian,s.等人,《有机化学杂志(j.org.chem.)》1963,28,2604)。在1966年,inuoye等人首次从链霉菌(streptomyces)菌株中分离出式(ii)的野尻霉素(nojirimycin)(inuoye等人《抗生素杂志(j.antibiot.)》1966,9,288),次年,paulsen等人报道了式(iii)的1-脱氧野尻霉素的首次合成(paulsen等人《化学报告(chem.ber.)》1967,100,802)。
从那时起,由于发现了亚氨基糖对糖苷酶的强抑制活性,对亚氨基糖的兴趣增加了。在生理ph值下,氮原子带正电,并且这允许了与存在于酶的活性位点中的末端羧酸根的相互作用。另外,亚氨基糖在结构上与自然界中存在的氧类似物相似。
然而,1-脱氧半乳糖野尻霉素在自然界中不存在,并且因此必须制备。paulsen等人(《化学报告》1980,113,2601)公开了从1,6-脱水-α-d-半乳呋喃糖开始的第一种合成途径。从那时起,已经公开了使用糖或糖类似物作为起始材料的1-脱氧半乳糖野尻霉素的各种合成程序。
barili等人(《四面体(tetrahedron)》1997,53,3407)已经报道了一种合成半乳糖构型的氮杂糖的有趣方法。作者描述了1-脱氧半乳糖野尻霉素的制备,其通过从式(iva)的l-阿拉伯-己-5-酮糖(l-arabino-hexos-5-ulose)开始、用二苯甲胺的双重还原胺化(流程1),以及随后的脱苄基步骤。发现双重还原胺化的关键反应步骤中的产率为36%。
流程1
然后将如此获得的d-半乳糖异构体通过色谱法纯化。同样在用不同胺的式(ivb)的双保护衍生物的情况下,最终产物也必须通过色谱法纯化。此外,所报道的程序使用nacnbh3作为还原剂,相对于式(iv)的化合物过量使用nacnbh3,具体而言,每摩尔式(iva)的l-阿拉伯-己-5-酮糖中使用2.2摩尔还原剂。当使用nacnbh3时必须采取重要的预防措施,以防止释放有毒并危险的hcn。此外,该文章没有提及如何处理含有氰化物离子的废水,这是使该方法在工业规模上可应用的主要问题。
因此,需要一种新的用于制备式(i)的米加司他的方法,该方法克服了上述缺点并且允许以有效方式在工业规模上、对人类健康和环境安全并且以高产率和高纯度(特别是立体化学纯度)获得所需产品。
附图和分析方法的简要说明
通过x射线粉末衍射(xrpd)表征呈结晶形式(在本文中称为形式i)的米加司他盐酸盐。xrpd谱图是在以下操作条件下用带有psdlynxeye检测器的自动粉末和液体衍射仪brukerd8advance收集的:布拉格-布伦塔诺几何(bragg-brentanogeometry),经镍滤波的cukd
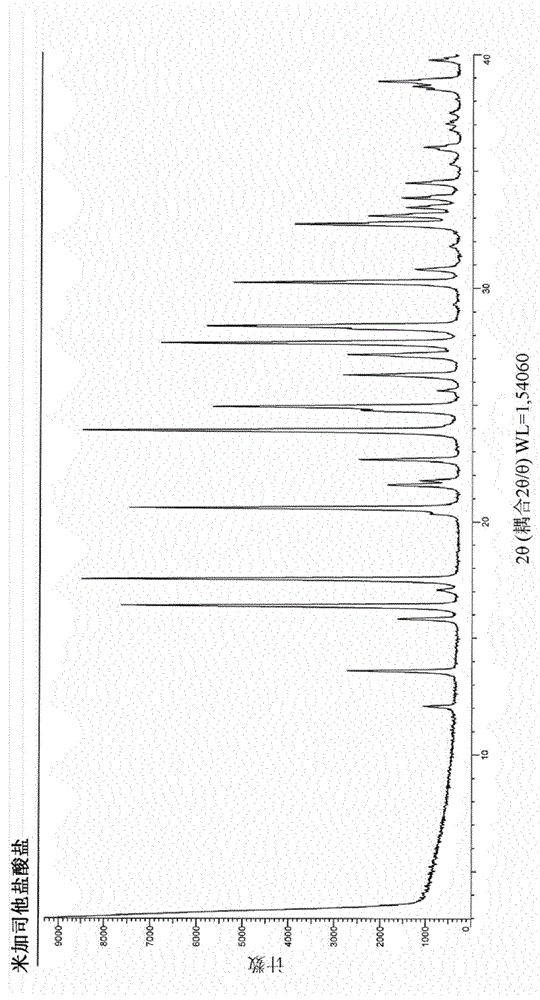
图1示出了呈结晶形式(在本文中称为形式i)的米加司他盐酸盐的xrpd图谱,其特征在于xrpd图在2θ中的以下处(在2θ中以°为单位表示):12.07;13.58;15.81;16.37;17.06;17.53;20.59;21.59;21.75;22.67;39.95;24.92;26.30;27.16;27.67;28.37;30.26;30.81;32.74;33.11;33.46、33.87和34.52±0.2°具有峰。
技术实现要素:
本发明的目的是用于制备式(v)化合物或其盐的方法
其中x彼此相等,是氢或醇保护基,
r是氢、c1-c6烷基或氨基保护基;
所述方法包含在还原剂和溶剂的存在下,式(vi)的二羰基化合物
其中每个x1彼此相等,是醇保护基,与胺nh2-r或其盐的双重还原胺化步骤,并且如果是这种情况的话,
将式(v)的化合物转化为另一种式(v)的化合物或其盐,和/或将式(v)的化合物的盐转化为其游离碱。
具体实施方式
本发明的目的是用于制备式(v)化合物或其盐的方法
其中x彼此相等,是氢或醇保护基,
r是氢、c1-c6烷基或氨基保护基;
所述方法包含在还原剂和溶剂的存在下,式(vi)的二羰基化合物
其中每个x1彼此相等,是醇保护基,与式(vii)的胺或其盐
nh2-r
(vii)
其中r如上定义,的双重还原胺化步骤,并且如果是这种情况的话,
将式(v)的化合物转化为另一种式(v)的化合物或其盐,和/或将式(v)的化合物的盐转化为其游离碱,以及如果是这种情况的话,
从其混合物中分离单一异构体。
醇保护基x和x1是例如从碳水化合物化学中已知的保护基,例如苄基、烯丙基、乙酰基或苯甲酰基。
优选的醇保护基是苄基。
c1-c6烷基,其可以是直链或支链的,通常是c1-c4烷基,如甲基、乙基、丙基、异丙基或丁基、异丁基、叔丁基。c1-c6烷基可以任选地被一个或多个取代基取代,所述取代基可以相同或不同,优选一至三个取代基,如羟基或卤素,特别是氯或氟。
氨基保护基是例如从肽化学中已知的保护基,例如苄基、二苯甲基、三苯甲基、1-苯乙基、苄氧羰基和叔丁氧羰基,优选苄基、二苯甲基、三苯甲基或1-苯乙基。
在式(vii)的胺或其盐中,r优选为苄基、二苯甲基、三苯甲基或1-苯乙基。在1-苯乙基的情况下,式(vii)的胺在绝对构型(s)((1s)-1-苯乙胺或(s)-phea)或(r)((1r)-1-苯乙胺或(r)-phea),或其立体异构体混合物的情况下可以是光学活性的。
优选的氨基保护基是苄基。
进一步优选的氨基保护基是二苯甲基。
进一步优选的氨基保护基是三苯甲基。
进一步优选的氨基保护基是1-苯乙基,其中对应的胺具有绝对构型(s)或(r)或其立体异构体混合物。
进一步优选的氨基保护基是1-苯乙基,其中对应的胺具有绝对构型(s)。
进一步优选的氨基保护基是1-苯乙基,其中对应的胺具有绝对构型(r)。
在还原胺化反应中使用的还原剂可以是例如nacnbh3。
在另一个实施例中,在还原胺化反应中使用的还原剂可以是例如nabh4。
在另一个实施例中,在还原胺化反应中使用的还原剂可以是例如nabh(oac)3。
在又一个实施例中,在还原胺化反应中使用的还原剂可以是例如式(viii)的硼烷与胺的复合物
bh3·ny3
(viii),
其中每个取代基y相同或不同,是氢、任选取代的c1-c6烷基或芳基,或两个y,连同与其相连的氮原子一起形成任选地含有氧原子或nra基团的c5-c6杂环基环,其中ra是氢、氨基保护基或c1-c4烷基。
在又一个实施例中,在还原胺化反应中使用的还原剂可以是例如式(ix)的硼烷与吡啶的复合物
其中每个w取代基相同或不同,是氢、任选取代的c1-c6烷基或卤素原子。
优选的式(viii)的胺是哌啶。
进一步优选的式(viii)的胺是吗啉。
进一步优选的式(viii)的胺,其中c5-c6杂环基环含有nra基团,是哌嗪,其中ra是氢或c1-c4烷基,并且其中c1-c4烷基优选是甲基、乙基或丙基。
芳基可以是例如苯基。苯基可以任选地被一至三个取代基取代,所述取代基可以相同或不同、独立地选自直链或支链c1-c4烷基,所述c1-c4烷基又可以任选被以下取代:一至三个卤素原子,通常是氟;羟基;c1-c4烷氧基,例如甲氧基;卤素原子,如溴或氯;氰基;和硝基。
式(viii)或式(ix)的还原剂是可商购的。
替代地,式(viii)或式(ix)的还原剂可以由与thf或me2s复合的硼烷溶液开始,通过用对应的式(viiia)的胺或对应的式(ixa)的吡啶处理来原位制备,其中y和w如上定义。
在本发明的优选方面,式(viiia)的胺为仲胺,更优选地一个y为h,并且其余的两个y连同与其相连的氮原子一起一起形成吗啉、哌啶或哌嗪环。优选地,式(viii)的还原剂是原位制备的。
适合于本发明的进一步的还原剂是与thf或与me2s复合的硼烷。这些还原剂是可商购的,或者可以根据已知方法制备成原位转变。
式(viii)或式(ix)的还原剂导致极其有效、经济、安全并且因此特别在工业水平上是有利的。类似地,与其他已知的双重还原胺化程序相比,从反应混合物中分离产物和废水的处理变得更容易。
还原胺化反应可以任选地在溶剂的存在下进行,所述溶剂选自例如偶极非质子溶剂,通常为二甲基甲酰胺、二甲基乙酰胺、乙腈或二甲基亚砜;醚,通常为四氢呋喃或二噁烷或甲基叔丁基醚;氯化溶剂,通常为二氯甲烷;非极性溶剂,通常为甲苯或己烷;酯,如乙酸乙酯、乙酸异丙酯或乙酸丁酯;极性质子溶剂,通常为c1-c4醇,优选甲醇或水,或所述溶剂中的两种或更多种(优选两种或三种)的混合物。
式(v)或式(vii)的化合物的盐通常是其药学上可接受的盐。
还原胺化可以通过用(每摩尔式(vi)的化合物)至少化学计算量的式(vii)的胺和至少2摩尔的式(viii)或(ix)的还原剂处理式(vi)的二羰基化合物来进行。
还原胺化可有利地在约-20℃至约40℃之间,优选在约0℃至约40℃之间的反应温度下,更优选在约25℃下并且通常在反应混合物的ph值在约8至约4之间的情况下进行。
反应混合物的ph值可以通过添加乙酸来调节。
式(vi)化合物的双重还原胺化步骤可能导致形成两种非对映异构体:
barili等人(《四面体》1997,53,3407)已经观察到两种非对映异构体的形成。作者用未保护的二羰基化物(式(iva)的化合物)和二苯甲胺进行了双重还原胺化,并且发现仅形成了d-半乳糖异构体,产率为36%。用式(ivb)的双保护二羰基化物进行还原胺化,得到70:30的d-半乳糖:l-阿卓糖非对映异构体的混合物,产率为38%。因此,进行从式(ivb)的双保护化合物开始的方法,以与式(iva)的未保护二羰基化物几乎相同的产率提供亚氨基糖。另外,部分保护导致两种非对映异构体的混合物,即使在用乙酰基保护基进一步保护游离羟基的情况下,通过薄层色谱法(tlc)也无法将其与几种洗脱系统分离。
本发明的发明人令人惊奇地发现,其中x1为苄基的式(vi)的四保护苄氧基化合物的反应不仅以显著更高的产率(68%)提供了亚氨基糖,而且还提供了更高量的d-半乳糖异构体。另外,两种非对映异构体都可以通过色谱法分离。
其中bn=苄基,并且r=二苯甲基
*游离羟基乙酰化后,所得产物只能纯化和分离为两种非对映异构体的混合物。产率是指双乙酰化的化合物。
本发明的发明人还令人惊讶地发现,还原剂和胺的选择对非对映异构混合物的形成有影响。
例如,用苄胺和nacnbh3作为还原剂的双重还原胺化产生的非对映异构混合物的d-半乳糖:l-阿卓糖比为2:1,而使用硼烷吗啉(式(viii)的本发明的还原剂)产生的非对映异构比d-半乳糖:l-阿卓糖为1:2。
根据本发明,式(vi)的四苄氧基化合物与(1s)-1-苯乙胺和硼烷吗啉的反应提供了的非对映体混合物d-半乳糖:l-阿卓糖为24:76,而用(1r)-1-苯乙胺和硼烷吗啉的方法提供了的非对映体混合物d-半乳糖苷:l-阿卓糖为80:20。
反应完成后,可以任选地用水洗涤混合物,并且可以用本领域技术人员已知的方法萃取式(v)的化合物。例如,可以通过结晶或色谱法来纯化任选地用水洗涤、用有机溶剂萃取的式(v)化合物。
在优选的实施例中,可以通过结晶来纯化混合物,包括从其混合物中分离d-半乳糖和/或l-阿卓糖异构体。
在另一个优选的实施例中,可以通过色谱法来纯化混合物,包括从其混合物中分离d-半乳糖和/或l-阿卓糖异构体。
非对映体混合物可以例如通过正相液相色谱法(nplc),例如通过正相快速柱色谱法,使用溶剂作为洗脱剂来纯化。纯化可以以等度或梯度洗脱模式进行。
优选的洗脱剂选自二氯甲烷、乙酸乙酯、己烷或丙酮,更优选己烷和乙酸乙酯的混合物,例如比率为9:1(v:v)。
任选地,可以根据已知方法将式(v)的化合物转化为其药学上可接受的盐。
本发明的另一个实施例是如上定义的用于制备式(v)的化合物的l-阿卓糖异构体的方法。
本发明的另一个实施例是如上定义的用于制备式(v)的化合物与其l-阿卓糖异构体的混合物的方法。
假如醇保护基为烯丙基,可以根据已知方法除去这类基团,例如通过异构化然后水解或在pd/c存在下氢化。
假如醇保护基为乙酰基或苯甲酰基,可以根据已知方法,例如通过在酸性条件或碱性条件下水解来进行除去。
在醇保护基为苄基的情况下,这类基团可以通过催化氢化除去。
假如氨基保护基为苄基、二苯甲基、苯乙基或苄氧羰基,保护基可以通过催化氢化除去。
假如氨基保护基为叔丁氧基羰基,可以通过用酸例如三氟乙酸或盐酸处理来除去保护基。
可以根据已知方法,例如通过催化氢化,通过除去保护基,将式(v)的化合物(其中r是氨基保护基,具体地苄基、二苯甲基、三苯甲基、1-苯乙基或苄氧羰基,并且每个x是醇保护基,具体地苄基)转化为另一种式(v)化合物(其中x和r是氢),即式(i)的米加司他或其盐。
可以根据已知方法,例如通过催化氢化,通过除去保护基,将式(v)的化合物(其中r是氢并且每个x是醇保护基,具体地苄基)转化为另一种式(v)的化合物(其中x和r是氢),即式(i)的米加司他或其盐。
催化氢化可以在均相或非均相金属催化剂,例如基于pd、pt、ni、rh或ru的催化剂的存在下进行。优选地,催化剂基于pd。
在非均相金属催化剂的情况下,优选将催化剂置于惰性载体上,所述惰性载体例如为碳、氢氧化钡、氧化铝、碳酸钙;优选木炭。载体上金属的浓度可在约1%和30%之间变化,优选在约5%和20%之间变化。
所使用的氢气压力可以在约1atm至40atm之间变化,优选地在约1atm至10atm之间。
相对于式(v)的化合物,所用催化剂的摩尔量在约0.1%和10%之间,优选在约0.5%和5%之间。
反应可以在有机溶剂的存在下进行,所述有机溶剂例如选自极性非质子溶剂,如二甲基甲酰胺、二甲基乙酰胺、乙腈或二甲基亚砜;无环或环状醚溶剂,通常为四氢呋喃或甲基叔丁基醚;氯化溶剂,通常为二氯甲烷;非极性非质子溶剂,例如甲苯或己烷;极性质子溶剂,例如直链或支链c1-c6醇,具体地甲醇、乙醇、异丙醇或丁醇;水;酯,例如乙酸乙酯、乙酸异丙酯或乙酸丁酯;直链或支链的c3-c7酮,例如丙酮、甲基乙基酮,或甲基异丁基酮;羧酸,例如乙酸或丙酸;或上述溶剂中两种或更多种(通常两种或三种)的混合物。
替代地,氢化反应可以在一种、两种或三种以上指示的有机溶剂(包含无机酸或碱,例如盐酸或硫酸,或氢氧化钠或氢氧化钾)的溶液或混合物中进行。
所述氢化反应可以在约0℃至溶剂或溶剂体系苄氧羰基的回流温度之间的温度下进行,优选在约25℃至回流温度之间的温度下进行。
本发明的另一个目的是用于制备式(i)的米加司他或其药学上可接受的盐的方法,包含使用根据本发明的方法获得的式(v)的化合物。
根据另一方面,本发明提供了呈结晶形式(在本文中称为形式i)的米加司他盐酸盐,其特征在于xrpd图在2θ中的以下处(在2θ中以°为单位表示):12.07;13.58;15.81;16.37;17.06;17.53;20.59;21.59;21.75;22.67;39.95;24.92;26.30;27.16;27.67;28.37;30.26;30.81;32.74;33.11;33.46、33.87和34.52±0.2°具有峰。
式(vi)的二羰基化合物的制备
其中每个基团x1彼此相同,是醇保护基,可以通过在氧化剂和任选地溶剂的存在下,式(x)的二醇的氧化来进行,
其中每个x1如上定义。
氧化反应可以通过本领域技术人员已知的方法进行,例如通过使用活化的二甲基亚砜(dmso)、基于铬的试剂(cro3·py2、pdc、pcc)、基于五价碘的氧化剂(戴斯-马丁高碘剂(dess-martinperiodinane)、ibx)、tempo或naocl的方法进行。优选地,氧化反应可以用活化的二甲基亚砜(dmso)进行。
氧化反应可通过用(每摩尔式(x)的化合物)至少化学计算量的活化dmso,例如用约2摩尔的dmso处理式(x)的化合物来进行。
dmso可以通过本领域技术人员已知的方法活化,例如使用活化剂,如草酰氯(cocl)2,在-78℃和-60℃之间范围内的温度下;氰尿酰氯或三氟乙酸酐,在-45℃和-30℃之间的温度下;py·so3或p2o5,在0℃和30℃之间的温度下。活化剂与式(x)化合物之间的摩尔比通常在约2:1和8:1之间,优选在约2:1和5:1之间。将反应混合物在适合于使试剂活化的温度下搅拌约10分钟至2小时。然后,加入碱,并使混合物反应足够的时间以完成氧化,通常在少于5小时内。碱可以是有机或无机碱。优选地,碱是有机碱,如叔胺,具体地三乙胺、二异丙基乙胺、二氮杂双环十一碳烯或二氮杂双环辛烷。
氧化反应可以任选地在溶剂的存在下进行,所述溶剂例如为偶极非质子溶剂,通常为二甲基甲酰胺、二甲基乙酰胺、乙腈、dmso;或醚,通常为四氢呋喃,或二噁烷或甲基叔丁基醚;氯化溶剂,通常为二氯甲烷;非极性溶剂,通常为甲苯或己烷,酯,例如乙酸乙酯、乙酸异丙酯或乙酸丁酯;极性质子溶剂,通常为c1-c4醇,优选甲醇,或水,或所述溶剂中的两种或更多种(优选两种或三种)的混合物;或以也作为反应溶剂的量使用二甲基亚砜。优选地,溶剂是dmso。
可以在反应结束时用本领域技术人员已知的方法从反应混合物中分离式(vi)的化合物。式(vi)的化合物,一旦在用水洗涤后萃取,优选原样用于随后的还原胺化反应中,得到式(v)的化合物,然后式(i)的米加司他或其药学上可接受的盐。
本发明的另一个目的是用于制备式(i)的米加司他或其药学上可接受的盐的方法,其包含使用式(v)或式(vi)的化合物。
式(x)的化合物可以按照例如《有机化学通讯(org.lett.)》2007,9,879中所报道的通过使式(xi)的化合物还原来制备,
其中每个x1如上定义。还原剂和任选的溶剂是本领域技术人员已知的。
式(xi)的化合物是d-半乳糖的衍生物,其是可商购的或可以通过本领域技术人员已知的方法,从半乳糖开始制备,例如在《日本化学学会公报(bull.chem.soc.jpn.)》1976,49(9),2639中所描述。
下列实例进一步说明本发明。
实例1:式(vi)的2,3,4,6-四-o-苄基-d-阿拉伯-己-5-酮糖(2,3,4,6-tetra-o-benzyl-d-arabino-hexos-5-ulose)的合成
在氮气气氛下将2.03ml(23.8mmol)的(cocl)2溶于5mlch2cl2中,并将溶液冷却至-75℃。在15分钟内,保持温度低于-60℃,滴加2.12ml(29.9mmol)的dmso在5mlch2cl2中的溶液,并将反应混合物再搅拌35分钟。然后在15分钟内,保持温度低于-60℃,将3.0g(5.5mmol)的式(x)的2,3,4,6-四-o-苄基-d-半乳糖醇(《有机化学通讯》2007,9,879)在10mlch2cl2中的溶液添加到混合物中,并将反应混合物再搅拌2小时。然后在20分钟内,保持温度低于-45℃,滴加7.78ml(55.9mmol)的三乙胺(tea)。然后,使式(vi)的2,3,4,6-四-o-苄基-d-阿拉伯-己-5-酮糖溶液达到室温,并且无需进一步纯化即可用于下一步。
实例2:式(v)的(2r,3s,4r,5s)-2-[(苯基甲氧基)甲基]-3,4,5-三-(苯基甲氧基)-哌啶的合成
在0℃下,将适当的式(vii)的胺(55.9mmol)、5ml(87.4mmol)的乙酸和0.9g(14.4mmol)的nacnbh3溶解在20ml的甲醇中。将如实例1中所述获得的式(vi)的2,3,4,6-四-o-苄基-d-阿拉伯-己-5-酮糖溶液添加到上述溶液中,并将反应混合物搅拌在0℃下搅拌1.5小时,然后使其达到室温。然后将混合物再次冷却至0℃,并缓慢添加10ml的h2o和10ml的30%naoh。添加30ml的ch2cl2,分离各相,并用ch2cl2进一步萃取水相。合并的有机相用20ml的3mhcl、30ml的11%naocl溶液、20ml的10%na2s2o3溶液、40ml的nahco3的饱和溶液和h2o(2×25ml)洗涤。有机相经无水硫酸钠干燥,过滤并在减压下蒸发溶剂。通过快速色谱法(己烷/乙酸乙酯9:1,v:v)纯化粗反应混合物,得到2种非对映体(d-半乳糖和l-阿卓糖)的混合物。
对于二苯甲胺,可以通过快速色谱法(己烷/乙酸乙酯9:1,v:v)分离两种非对映异构体
实例3:barili等人(《四面体》1997,53,3407)的程序与本发明的方法的比较
本发明的作者已经应用了barili等人(《四面体》1997,53,3407)的反应条件,对于式(vi)的化合物,其中x1是苄基,使用二苯甲胺和nacnbh3作为还原剂。令人惊讶的是,以66%的产率获得式(v)的产物,该产率远高于报道的对于式(iva)的未保护二羰基化物(其中p=h)的36%。其中p=bn的式(ivb)的2,6-二苄基-二羰基的还原胺化和随后的乙酰化步骤提供了3,4-o-乙酰基-n-二苯甲胺d-半乳糖和l-阿卓糖衍生物的混合物70:30,产率为38%。然后用甲醇钠的甲醇溶液将乙酰基脱保护,产率为92%。
实例4:式(v)的(2r,3s,4r,5s)-2-[(苯基甲氧基)甲基]-3,4,5-三-(苯基甲氧基)-哌啶的合成
在0℃下,将适当的式(vii)的胺(55.9mmol)、乙酸(5ml,87.4mmol)和硼烷吗啉(1.45g,14.4mmol)溶解在20ml的甲醇中。将如实例1中所述制备的式(vi)的化合物的溶液添加到混合物中,将其在0℃下搅拌1.5小时,然后使其在夜间达到室温。然后将混合物冷却至0℃,并缓慢添加h2o(10ml)和30%naoh(10ml)。添加30ml的ch2cl2,分离各相,并用ch2cl2萃取水相。合并有机相,并用3mhcl、11%naocl溶液、10%na2s2o3溶液、nahco3饱和溶液洗涤,并且最后用水洗涤。有机相经无水硫酸钠干燥,过滤并在减压下蒸发溶剂。通过快速色谱法(己烷/乙酸乙酯9:1,v:v)纯化粗反应混合物,得到2种绝对构型的非对映体的混合物,分别为d-半乳糖和l-阿卓糖。
实例5:式(i)的米加司他的合成
在氮气气氛下,将1.95g(2.83mmol)的式(v)的(2r,3s,4r,5s)-1-二苯甲基-2-[(苯基甲氧基)甲基]-3,4,5-三-(苯基甲氧基)-哌啶)溶于20mlmeoh和1ml6mhcl的混合物中。添加300mgpd/c20%,并将悬浮液在氢气气氛下于室温下搅拌。反应结束时,将悬浮液在珍珠岩板上过滤并浓缩。将获得的固体在meoh中搅拌,然后通过布氏漏斗过滤并用甲醇洗涤。在50℃下真空干燥后,获得429mg呈结晶形式(在本文中称为形式i)的米加司他盐酸盐,并且其特征在于xrpd图在2θ中的以下处(在2θ中以°为单位表示):12.07;13.58;15.81;16.37;17.06;17.53;20.59;21.59;21.75;22.67;39.95;24.92;26.30;27.16;27.67;28.37;30.26;30.81;32.74;33.11;33.46、33.87和34.52±0.2°具有峰,产率为76%并且纯度高于98%。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种丙辛吲哚盐酸盐药物中间体丙辛吲哚的合成方法与制造工艺
- 一种吲哚洛尔药物中间体(一)‑4‑(2‑羟基‑3‑异丙氨基丙氧)吲哚的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺