一种拉链式可延伸探针在cfDNA文库构建中的应用的制作方法
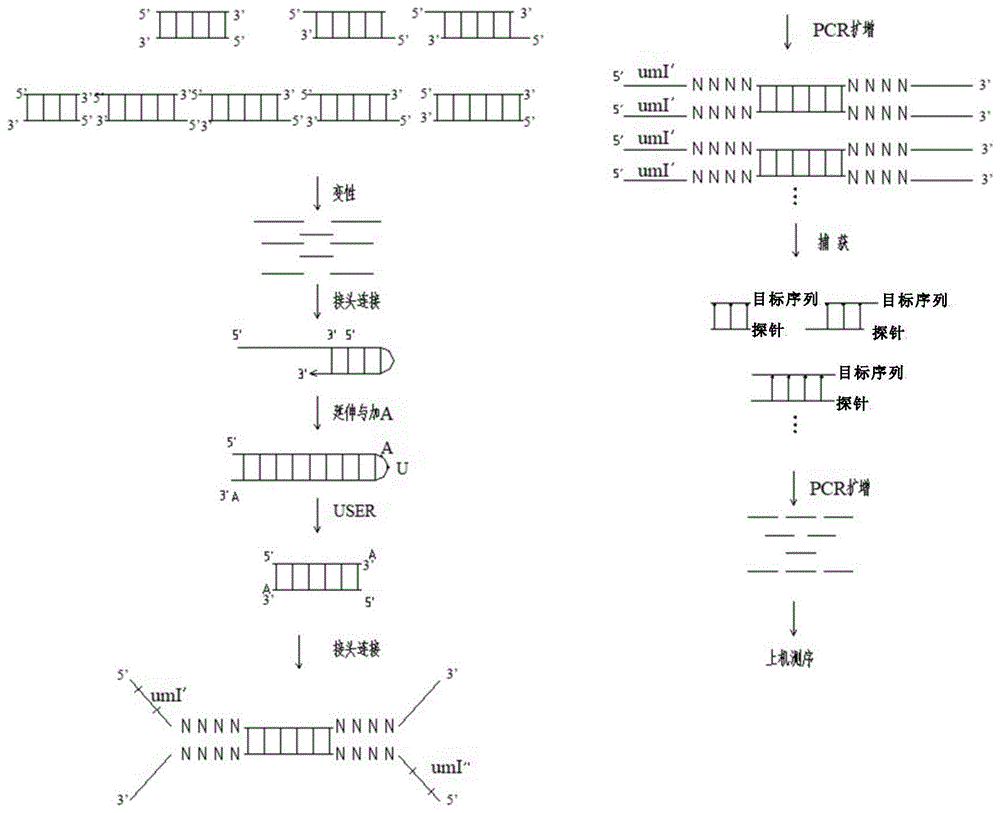
本发明属于生物技术,更具体地,本发明提供了一种拉链式可延伸探针在cfdna文库构建中的应用。
背景技术:
在标准的大规模平行测序中,很难检测到丰度低于2%的突变。要想检测到这些低频突变,需要检测到大量的读段数,这样会直接给测序成本带来极大地提升。这正是目前对血液中ctdna检测的瓶颈所在。1ml血浆中有1,800-13,000个cfdna片段,然而ctdna占有cfdna含量在0.4-11%。仅通过常规建库测序方法会使ctdna检测信号完全淹没在背景噪声中,其原因在于,在通常建库过程中,在对末端cfdna修复的过程中,会在内切酶的作用下切掉3’端序列。并且cfdna片段化过程不像基因组打断那样随机,如果突变位置发生在cfdna的3’的近端,按照常规的建库方法,将无法检测到突变位点信息。
技术实现要素:
在第一方面,本发明提供了一种使用拉链式可延伸探针的cfdna文库构建方法,所述方法包括:
(1)获得拉链式探针,所述拉链式探针的3’端包括与目标dna单链互补配对的15-20个碱基,优先选择16个碱基,5’端包括颈环结构,所述颈环结构中互补配对的碱基数目为18-50个,优选20个,所述颈环结构的环部从5’-3’方向包括a和u碱基;
(2)将dna样品打断成片段,变性成单链dna;
(3)所述拉链式探针与变性后的单链dna进行连接反应,将连接产物进行延伸,并在延伸链的3’端加上a,形成末端封闭的双链dna复合物;
(4)将双链dna复合物剪切,形成平末端3’都带有a的双链dna;
(5)将所述双链dna与接头连接;
(6)对连接接头的所述双链dna进行探针捕获;
(7)对所述捕获序列进行测序,获得捕获序列的测序结果。
在一个实施方案中,在(3)中在klenow片段的作用下进行延伸。
在一个实施方案中,在(4)中在user酶的作用下进行剪切。
在一个实施方案中,在(5)中所述接头包括分子标签。
在一个实施方案中,在(1)中所述拉链式探针的序列如下:从5’-3’为颈环部分序列+au+与颈环部分互补序列+与目标区域互补序列:
acggtattga++au+tcaataccgt+与探针互补序列。
在一个实施方案中,在(6)中包括对所述捕获产物测序的步骤。
在第二方面,本发明还提供了一种拉链式探针,所述拉链式探针的3’端包括与目标dna单链互补配对的15-20个碱基,优先选择16个碱基,5’端包括颈环结构,所述颈环结构中互补配对的碱基数目为18-50个,优选20个,所述颈环结构的环部从5’-3’方向包括a和u碱基。
在一个实施方案中,拉链式探针序列从5’-3’为颈环部分序列+au+与颈环部分互补序列+与目标区域互补序列:
acggtattga++au+tcaataccgt+与探针互补序列。
为了克服这个问题,发明人提出了单链转化为双链策略,然后对双链进行文库构建的方案,此方法最大的优势在于可以最大限度地保留cfdna中原始突变信息,可以使dna两条链中的每一条链同时被转化为用于上机测序的文库,有效地解决低频检测中遇到的瓶颈问题。
附图说明
通过以下附图对本发明进行说明
图1是本申请的方法的实验流程
图2示出了基因组打断与cfdna片段中同一突变点位置分布的异同。
图3单链转双链建库和双链建库比较的结果。
具体实施方式
图1是本申请的方法的实验流程。
图2示出了基因组打断与cfdna片段中同一突变点位置分布的异同:a图代表基因打断后n种情况下某一突变点位置在片段上的分布情况,由于在建库过程中,延伸酶都是由5’向3’端进行延伸,所以片段a片段上带有3’粘性末端会被限制性内切酶切掉,同时位于3’粘性末端的突变碱基也会被切掉,无法进入测序环节,因此无法被检测到。但是由于基因组打断的随机性,b、c、d、e等随机打断的片段上带有此突变点的信息不会被限制性内切酶切掉,因此会通过其它片段的补偿来弥补被限制性内切酶切掉的突变点信息。b图代表血浆中游离dna片段几种情况下的同一突变点位置的分布情况。在b图中,由于片段b’片段上带有3’粘性末端会被限制性内切酶切掉,同时位于3’粘性末端的突变碱基也会被切掉,无法进入测序环节,因此无法被检测到。虽然理论上通过a’和c’片段可以检测到突变点信息,但是受制于体内酶作用与cfdna断裂方式的有限性,b’片段的这种断裂方式可能会占比很高,另外建库过程中还会受制于酶转化效率的影响,因此有可能通过a’和c’片段的补偿,可能无法检测到突变位点信息。然而此突变位点信息可能对临床用药具有重大的指导意义,所以寻找一种可以检测到cfdna中突变位点信息显得尤为重要。
实施例
1.设计一段寡核苷酸链,这条链包括:3’端有15-20个碱基与目标dna单链互补配对,优先选择16个碱基,5’端有一段颈环结构,互补配对的碱基数目为18-50个,优选20个。在颈环的端部由5’-3’加入a和u碱基,加入a的目的是用于后续建库过程中的接头连接反应,加入u的目的是作为userenzyme酶切识别位点。在t4dna连接酶的作用下,拉链式接头与变性后的单链dna进行连接反应,连接产物在klenowfragment的作用下进行延伸并在延伸链的3’端加上a,形成末端封闭的双链dna复合物,在userenzyme酶的作用下进行剪切,转化为平末端3’带都带有a的双链dna,用于后续的普通双链建库的连接反应中。
拉链式接头序列设计如下:
5’-3’方向:
颈环部分序列+au+与颈环部分互补序列+与目标区域互补序列:
acggtattga++au+tcaataccgt+与探针互补序列(16)
2.拉链式接头退火:
拉链式接头变性5min之后,立即把制备有接头的管子置于冰上10min。
3.转化cfdna为单链
3.1将提取后的cfdna完全转移至pcr管中,按如下表格配置反应体系
3.2使用移液器上下吹打混匀,将pcr管置于水浴锅,37℃孵育10min。
3.395℃变性5min,立即转移pcr反应管至冰上,至少1min。
4.连接反应
4.1使用移液器上下吹打混匀,将pcr管置于室温孵育10min。
4.2用qiaquickpcr纯化试剂盒对上述产物进行纯化,洗脱体积为35ul。
5.延伸并加a和酶切反应
5.1使用移液器上下吹打混匀,将pcr管置于水浴锅,37℃孵育30min。
5.2用qiaquickpcr纯化试剂盒对上述产物进行纯化,洗脱体积为35ul。
6.加带有分子标签的接头
6.1接头连接
hybblock指的是封闭接头和高拷贝dna片段的序列,封闭接头的序列为:
adapter-1:aatgatacggcgaccaccgagatctacac[分子标签]acactctttccctacacgacgctcttccgatct
adapter-2:gatcggaagagcacacgtctgaactccagtcac[index]atctcgtatgccgtcttctgcttg
1)轻轻吸打混匀6次,避免产生气泡,然后短暂离心;
2)运行pcr仪程序(要求无热盖),设置pcr仪参数,加热模块设置20℃,时间15min。将pcr管放入pcr仪,运行程序;
6.2磁珠纯化
1)将ampurexp磁珠在室温提前震荡混合均匀,室温孵育30min备用;
2)在步骤3进行完的pcr管中,按照0.8x体积的纯化磁珠进行实验;
3)轻轻吸打混匀6次;
6)室温静置孵育5min,将pcr管置于磁力架上3min;
7)移除上清,pcr管继续放置在磁力架上,向pcr管内加入40μl缓冲液a,涡旋混匀,放置5min;
4)将pcr管置于磁力架上,3min移除上清,向pcr管内加入200μl,80%乙醇溶液,静置30s后彻底移除上清。
5)将pcr管继续放置在磁力架上,向pcr管内加入200μl80%乙醇溶液,静置30s后彻底移除上清。
6)室温静置3-5min,使残留乙醇彻底挥发;
7)加入22ul的重悬nfh2o,把pcr管从磁力架取下,轻轻吸打重悬磁珠,避免产生气泡,室温静置5min;
8)将pcr管置于磁力架上2min;
9)用移液器吸取20μl上清液,用于下一步的pcr扩增。
7.pre-pcr反应
p5:aatgatacggcgaccaccga
p7:caagcagaagacggcatacga
1)使用移液器轻轻吹混匀,然后短暂离心;
2)将样品置于pcr仪上,启动pcr程序,如下:
热盖100℃
95℃维持45s
12个循环
98℃维持15s
60℃维持30s
72℃维持30s
72℃维持1min
4℃保持
8.pcr扩增后纯化
1)向pcr产物中加入50μlampurexp磁珠,用移液器吹打混匀,避免产生气泡;
2)室温孵育5min,把pcr管置于磁力架上3min;
3)移除上清,pcr管继续放置在磁力架上,向pcr管内加入200μl80%乙醇溶液,静置30s;
4)移除上清,再向pcr管内加入200μl80%乙醇溶液,静置30s后彻底移除上清。
5)室温静置5min,使残留乙醇彻底挥发;
6)加入25ulnfh2o,将离心管从磁力架取下,使用移液器,轻轻吸打重悬磁珠;
7)室温静置5min,将200μlpcr管置于磁力架上2min;
8)用移液器将上清液转移到新的200μlpcr管中(置于冰盒上),在反应管上标记好样本号,准备下一步反应;
9.样品、探针杂交
按照以下体系将样品文库与hybblock混匀标记为b
hybblock指的是封闭接头和高拷贝dna片段的序列,封闭接头的序
列为:
adapter-1-1的封闭序列为:
agatcggaagagcgtcgtgtagggaaagagtgt[分子标签]gtgtagatctcggtggtcgccgtatcatt
adapter-2-2的封闭序列:
caagcagaagacggcatacgagat[index]gtgactggagttcagacgtgtgctcttccgatc
封闭高拷贝dna片段的序列为打断的人类基因组序列cot-1。
1)将准备好的样品和hybblock混合物放入真空浓缩离心机,打开pcr管盖,启动离心机,打开真空泵开关,开始浓缩;
2)将抽干的样品重新溶在9ulnfh2o中待用;
3)将杂交缓冲液置于室温融化,未加热前会有沉淀出现,混匀后置于65℃水浴锅内预热,完全溶解后取20ul杂交缓冲液置于新的200μlpcr管内,盖好管盖,标记为a,继续置于65℃水浴锅内孵育待用;
4)取5ulrnaseblock与2ulprobe置于200μlpcr管内,轻轻吸打混匀,短暂离心后置于冰上待用,标记为c;
5)设置pcr仪参数,热盖100℃,95℃维持5min;65℃保持;
6)将pcr管b置于pcr仪上,运行以上程序;
7)pcr仪温度降至65℃时,将pcr管a置于pcr仪上孵育,盖上pcr仪热盖;
8)5min后,将c置于pcr上孵育,盖上pcr仪热盖;
9)将pcr管c放置入pcr仪2min后,把移液器调至13ul,从pcr管a中吸取13ul杂交缓冲液移至pcr管c中,吸取全部pcr管b中样品移至pcr管c中,轻轻吸打10次,充分混匀,避免产生大量气泡,密封管盖,盖上pcr仪热盖,65℃孵育过夜(8-16h)。
10.捕获磁珠准备
1)将磁珠(dynabeadsmyonestreptavidint1磁珠)从4℃取出,涡旋震荡重悬;
2)取50ul磁珠置于新的pcr管内,置于磁力架上1min,移除上清;
3)从磁力架上取下pcr管,加入200μl结合缓冲液轻轻吸打数次混匀,重悬磁珠;
4)置磁力架上1min,移除上清;
5)重复步骤3-4两次,共清洗磁珠3次;
6)从磁力架上取下pcr管,加入200μl结合缓冲液轻轻吸打6次重悬磁珠待用。
11.捕获目标区域dna文库
1)保持杂交产物pcr管c在pcr仪上,将步骤10中第6)步重悬后的200μlmyonet1磁珠加入到杂交后的产物pcr管c中,用移液器吸打6次混匀,置于旋转混匀仪上室温结合30min;
2)将pcr管置于磁力架上2min,移除上清液;
3)向pcr管c内加入200μl的洗涤缓冲液1,轻轻吸打6次混匀,置于旋转混匀仪上清洗15min,然后短暂离心,将pcr管放于磁力架上2min,移除上清;
4)加入200ul的65℃预热的洗涤缓冲液2,涡旋混匀5s,置于thermomixer上65℃孵育10min,转速800转/min进行清洗;
5)短暂离心,将pcr管放于磁力架上2min,移除上清。重复洗2次清洗,共计3次。最后一次彻底移除洗涤缓冲液2(可用10ul移液器移除残留);
6)pcr管继续置于磁力架上,向pcr管内加入200ul80%乙醇,静置30s后彻底移除乙醇溶液(可用10ul移液器移除残留),室温晾干2min;
7)向pcr管加入20μl无核酸酶水,从磁力架上取下pcr管,轻轻吸打6次重悬磁珠待用。
12.post-pcr反应
1)捕获后需要对dna文库进行富集,根据下表配制混合物
p5:aatgatacggcgaccaccga
p7:caagcagaagacggcatacga
2)将移液器调至35ul,轻轻吸打混匀6次,然后置于pcr仪上。
3)运行pcr仪程序:
热盖100℃
95℃维持4min
15个循环
98℃维持20s
65℃维持30s
72℃维持30s
72℃维持5min
12℃保持
4)pcr结束后向样品加入45μlagencourtampurexp磁珠,用移液器轻轻吸打6次混匀;
5)室温孵育5min,把pcr管置于磁力架上3min;
6)移除上清,pcr管继续置于磁力架上,加入200μl80%无水乙醇,静置30s;
7)移除上清,再向pcr管内加入200μl80%无水乙醇,静置30后彻底移除上清(可用10ul移液器移除底部残留酒精);
8)室温放置5min,使得残留乙醇彻底挥发;
9)加入25ulnuclease-freewater,将pcr管从磁力架拿下,轻轻吹打混匀重悬磁珠,室温放置2min;
10)将pcr管置于磁力架上2min;
11)用移液器吸23μl上清液转移到1.5ml离心管,标记样品信息。
实验结果及分析:
统计egfrl858r突变位点距离3’碱基数为10-50bp分别占有的读段数,结果如下:
如图3所示,根据二代测序结果进行分析统计,结果表明通过单链转化为双链建库方法得到的egfrl858r突变位点距离3’的碱基数为20-30bp的读段数最多,为2500条,而直接通过双链建库方法得到的egfrl858r突变位点距离3’的碱基数为10-20bp的读段数最多,为300条。双链建库方法得到的读段数随着距离3’碱基数目的增加,读段逐渐减少。如果突变位点距离3’的碱基个数较多,通过双链建库的方法会损失掉这些有意义的突变位点信息。同时通过单链转双链进行建库,会在很大程度上检测到距离3’不同碱基数目的读段数,在很大程度上提高了低频检测的灵敏度。尤其像egfrl858r这样的肿瘤耐药位点的检出,直接决定了肿瘤医生用药策略的改变。
序列表
<110>艾吉泰康生物科技(北京)有限公司
<120>一种拉链式可延伸探针在cfdna文库构建中的应用
<130>cf190086s
<160>7
<170>siposequencelisting1.0
<210>1
<211>22
<212>dna
<213>人工序列(syntheticsequence)
<400>1
acggtattgaattcaataccgt22
<210>2
<211>62
<212>dna
<213>人工序列(syntheticsequence)
<400>2
aatgatacggcgaccaccgagatctacacacactctttccctacacgacgctcttccgat60
ct62
<210>3
<211>57
<212>dna
<213>人工序列(syntheticsequence)
<400>3
gatcggaagagcacacgtctgaactccagtcacatctcgtatgccgtcttctgcttg57
<210>4
<211>20
<212>dna
<213>人工序列(syntheticsequence)
<400>4
aatgatacggcgaccaccga20
<210>5
<211>21
<212>dna
<213>人工序列(syntheticsequence)
<400>5
caagcagaagacggcatacga21
<210>6
<211>62
<212>dna
<213>人工序列(syntheticsequence)
<400>6
agatcggaagagcgtcgtgtagggaaagagtgtgtgtagatctcggtggtcgccgtatca60
tt62
<210>7
<211>57
<212>dna
<213>人工序列(syntheticsequence)
<400>7
caagcagaagacggcatacgagatgtgactggagttcagacgtgtgctcttccgatc57
- 还没有人留言评论。精彩留言会获得点赞!