荧光探针化合物及制备方法及其检测内毒素的应用和方法与流程
本发明涉及内毒素检测技术领域,尤其是涉及一种荧光探针化合物及制备方法及其检测内毒素的应用和方法。
背景技术:
细菌内毒素广泛存在于自然界中,例如,自来水中的内毒素含量为1-100eu/ml。当内毒素通过消化道进入人体时并不会产生毒性,但如果内毒素通过注射等方式进入血液则会引起不同的疾病。内毒素小量入血后可被肝脏枯否细胞灭活,不造成机体受损。内毒素大量进入血液就会引起发热反应,严重时甚至会导致死亡。因此,生物制品类、注射用药剂、化学药品类、放射性药物、抗生素类、疫苗类、透析液等制剂以及医疗器材类(如一次性注射器,植入性生物材料)必须在细菌内毒素检测试验合格后才能使用。
早期,最常用的热原(内毒素)检测方法是家兔法。后来,随着生物技术的不断发展,细菌内毒素试验(bacterialendotoxinstest,bet)逐渐替代了家兔热原试验,成为细菌内毒素检测最常用的方法。bet法即利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的含量是否符合规定的一种方法。这里所说的“鲎试剂”来源于海洋生物“鲎”的蓝色血液,它能够准确、快速地定性或定量检测样品中是否含有细菌内毒素。bet法可分为两种,即凝胶法和光度测定法,后者又包括浊度法和显色基质法。供试品检测时,可使用其中任何一种方法进行试验。当测定结果有争议时,除另有规定外,应以凝胶限度试验结果为准。
凝胶法系通过鲎试剂与内毒素产生凝集反应的原理进行限度检测或半定量检测内毒素的方法。
浊度法系利用检测鲎试剂与内毒素反应过程中的浊度变化而测定内毒素含量的方法。根据检测原理,可分为终点浊度法和动态浊度法。终点浊度法是依据反应混合物中的内毒素含量和其在孵育终止时的浊度(吸光度或透光率)之间存在的量化关系来测定内毒素含量的方法。动态浊度法是检测反应混合物的浊度到达某一预先设定的吸光度或透光率所需要的反应时间,或是检测浊度增加速度的方法。
显色基质法系利用检测鲎试剂与内毒素反应过程中产生的凝固酶使特定底物释放出呈色团的多少而测定内毒素含量的方法。根据检测原理,分为终点显色法和动态显色法。终点显色法是依据反应混合物中内毒素含量和其在孵育终止时释放出的呈色团的量之间存在的量化关系来测定内毒素含量的方法。动态显色法是检测反应混合物的吸光度或透光率达到某一预先设定的检测值所需要的反应时间,或检测值增加速度的方法。
鲎试剂虽然是检测内毒素较好的试剂,但其需要采集鲎的血液,对海洋生态环境造成影响,同时受限于鲎繁殖能力低下的影响,进而影响产能,且鲎试剂的质量受限于鲎的血液质量,难以把控。
因此中国专利cn105623645b公开了一种基于聚集诱导发光原理的荧光探针及其制备方法、应用和检测内毒素方法,其荧光探针3-乙基-2-(4-1,2,2-三苯基乙烯)烯基-苯并噻唑碘盐(tpe-be-i)能通过人工合成的方式进行生产,摆脱了鲎的制约。
但这种荧光探针利用的是荧光探针的聚集诱导发光(aie)特性和去碘离子荧光淬灭效应来检测内毒素的。tpe-be-i探针在dmso/水混合溶液中形成聚集体,理论上应该会发射出强烈的荧光,然而,由于结构中含有碘离子,会淬灭tpe-be-i聚集体的荧光,因此tpe-be-i聚集体未能表现出荧光特性。当待测物中含有内毒素(lps)时,内毒素(lps)中的磷酸负离子会将tpe-be-i聚集体中的碘离子置换出来,从而去除碘离子的荧光淬灭效应,使得检测体系荧光增强。
因此,当待测体系中含有能置换tpe-be-i中碘离子的成分时(例如,生理盐水和ringer’液),即使没有lps,检测体系的荧光也会增强,由此造成假阳性的结果,导致该荧光探针只能用于葡萄糖注射液、hepes缓冲液、超纯水或其它不含有碘离子置换成分的样品,因此,还有改善空间。
技术实现要素:
针对现有技术存在的不足,本发明的第一目的在于提供一种荧光探针化合物,其能适用于检测含有碘离子置换成分的样品,应用范围更广,填补了目前的技术空白。
为实现上述目的,本发明提供了如下技术方案:
一种荧光探针化合物,所述荧光探针化合物为4-(4-(1,2,2-三苯基乙烯基)苯基)乙基吡啶碘盐,分子式为c33h28ni,结构式为:
通过采用上述技术方案,荧光探针化合物为4-(4-(1,2,2-三苯基乙烯基)苯基)乙基吡啶碘盐(以下简称tpepye);利用荧光探针的聚集诱导发光(aie)特性、静电相互作用和亲疏水相互作用来检测内毒素,tpepye探针具有较好的水溶性,其在无热原水或检测体系中能充分溶解,不发射荧光;当待测物中含有内毒素(lps)时,lps中的磷酸负离子会将tpepye聚集体中的碘离子置换出来,从而去除碘离子的荧光淬灭效应;同时,tpepye探针与lps分子正负电荷之间相互吸引,且lps分子含有较多的疏水长烷基链,受静电相互作用和亲疏水相互作用的驱动,tpepye探针会与lps分子形成聚集体,发射出强烈的荧光;当待测体系中含有能置换碘离子的成分时,由于tpepye具有较好的水溶性,在不含有lps分子的溶液中不能形成聚集体,因此不会发射出荧光,从而可以排除碘离子置换物对检测结果的干扰;使得荧光探针化合物可以用于类似于氯化钠注射液(生理盐水)和盐酸氨溴索等含有可置换碘离子的氯离子的注射液中内毒素含量的检测,具有较好的抗干扰性,使得荧光探针化合物能适用于检测含有碘离子置换成分的样品,应用范围更广,填补了目前的技术空白;本发明的荧光探针化合物对内毒素的检测范围覆盖0.05-10eu/ml,而《中国药典》中对需要进行内毒素检测的药品的内毒素限值一般在0.1-5eu/ml,因此本发明的荧光探针化合物较为符合实际应用要求。
针对现有技术存在的不足,本发明的第二目的在于提供一种荧光探针化合物的制备方法,其包括以下步骤:
s1.取二苯基甲烷与4-溴二苯甲酮溶于有机溶剂中,在正丁基锂的作用下,反应得第一中间体;
s2.第一中间体与对甲苯磺酸溶于有机溶剂中升温至回流,反应得第二中间体;
s3.第二中间体与4-硼酸吡啶溶于有机溶剂中,在碳酸钾水溶液、四丁基溴化铵以及四(三苯基磷)钯的作用下,反应得第三中间体;
s4.将第三中间体与碘乙烷溶于有机溶剂中,搅拌反应得4-(4-(1,2,2-三苯基乙烯基)苯基)乙基吡啶碘盐。
通过采用上述技术方案,制备得到4-(4-(1,2,2-三苯基乙烯基)苯基)乙基吡啶碘盐(tpepye),tpepye具有较好的水溶性,当待测体系中含有能置换碘离子的成分时,在不含有lps分子的溶液中不能形成聚集体,因此不会发射出荧光,从而可以排除碘离子置换物对检测的干扰;使得荧光探针化合物可以用于氯化钠注射液(生理盐水)和盐酸氨溴索等含有可置换碘离子的氯离子的注射液中内毒素含量的检测,且不会造成干扰,具有较好的抗干扰性,使得荧光探针化合物的适用于检测含有碘离子置换成分的样品。
本发明进一步设置为:所述步骤s1在0℃至4℃的温度下进行反应。
通过采用上述技术方案,在0℃至4℃的温度下,正丁基锂比较稳定,不会因为活性高而产生危险。
本发明进一步设置为:所述步骤s3在75℃至85℃的温度下进行反应。
通过采用上述技术方案,在75℃至85℃的温度下,四(三苯基磷)钯的催化作用较佳,使得反应速率加快。
本发明进一步设置为:所述步骤s4在75℃至85℃的温度下进行反应。
通过采用上述技术方案,在75℃至85℃的温度下,第三中间体与碘乙烷的活性较高,易于反应,使得反应速率加快。
针对现有技术存在的不足,本发明的第三目的在于提供一种荧光探针化合物的应用,其可以检测能将荧光探针化合物中的碘离子置换出来的溶液中的内毒素浓度;
能将碘离子置换的溶液包括生理盐水、ringer’液。
通过采用上述技术方案,生理盐水、ringer’液中含有能置换碘离子的成分,由于tpepye具有较好的水溶性,在不含有lps分子的溶液中不能形成聚集体,因此不会发射出荧光,从而可以排除碘离子置换物对检测的干扰;使得荧光探针化合物可以用于氯化钠注射液(生理盐水)和盐酸氨溴索等含有可置换碘离子的氯离子的注射液中内毒素含量的检测,且不会造成干扰,具有较好的抗干扰性,使得荧光探针化合物的适用于检测含有碘离子置换成分的样品。
针对现有技术存在的不足,本发明的第四目的在于提供一种应用荧光探针化合物检测内毒素的方法,其包括以下步骤:
(1)将荧光探针化合物溶解于无热原水中,制备储存液;
(2)将内毒素标准品加入内毒素含量为0eu/ml且与待测溶液同种类的溶液样品中制备标准品溶液;
(3)取储存液与标准品溶液混合均匀得标准反应液,取储存液与待测溶液混合均匀得待测反应液;
(4)检测标准反应液得标准荧光曲线,检测待测反应液得待测荧光曲线;
(5)对比待测荧光曲线与标准荧光曲线得出待测溶液中的内毒素浓度是否达标。
通过采用上述技术方案,通过制备标准反应液以及待测反应液并检测出标准荧光曲线与待测荧光曲线,以通过对比标准荧光曲线与待测荧光曲线以判断待测溶液中的内毒素含量是否达标,使得操作十分方便,且根据不同待测溶液在《中国药典》中的限值以配制标准品溶液,使得通过标准品溶液得出的标准荧光曲线作为是否达标的标准荧光曲线时更为直观且结论可靠。
本发明进一步设置为:所述步骤(1)中存储液中荧光探针化合物的浓度为100-200μm。
通过采用上述技术方案,保证较好的荧光强度以保证荧光曲线清晰,保证结论可靠,同时避免荧光强度过高影响观察,并且控制成本,易于推广;在相同的内毒素含量下,探针的浓度越高,产生的聚集体越多,荧光信号强度越强,使得检测灵敏度较高。
本发明进一步设置为:所述步骤(3)中取储存液与若干不同内毒素含量的标准品溶液混合均匀得不同内毒素含量的标准反应液。
通过采用上述技术方案,通过制备若干不同内毒素含量的标准品溶液以制备出不同内毒素含量的标准反应液,以获得若干条不同内毒素含量的标准荧光曲线,使得对比待测荧光曲线时能更为直观的判断待测溶液中内毒素含量的数值所处的数值范围。
本发明进一步设置为:所述步骤(4)中用荧光分光光度计检测标准反应液以获得标准荧光曲线,用荧光分光光度计检测待测反应液以获得待测荧光曲线。
通过采用上述技术方案,通过荧光分光光度计检测得出标准荧光曲线以及待测荧光曲线,使得标准荧光曲线以及待测荧光曲线的结果更为准确可靠,且操作方便。
综上所述,本发明具有以下有益效果:
1.当待测体系中含有能置换碘离子的成分时,由于tpepye具有较好的水溶性,在不含有lps分子的溶液中不能形成聚集体,因此不会发射出荧光,从而可以排除碘离子置换物对检测的干扰;
2.荧光探针化合物对内毒素的检测范围覆盖0.05-10eu/ml,而《中国药典》中对需要进行内毒素检测的药品的内毒素限值一般在0.1-5eu/ml,因此荧光探针化合物较为符合实际应用要求;
3.通过对比标准荧光曲线与待测荧光曲线以判断待测溶液中的内毒素含量是否达标,使得操作十分方便。
附图说明
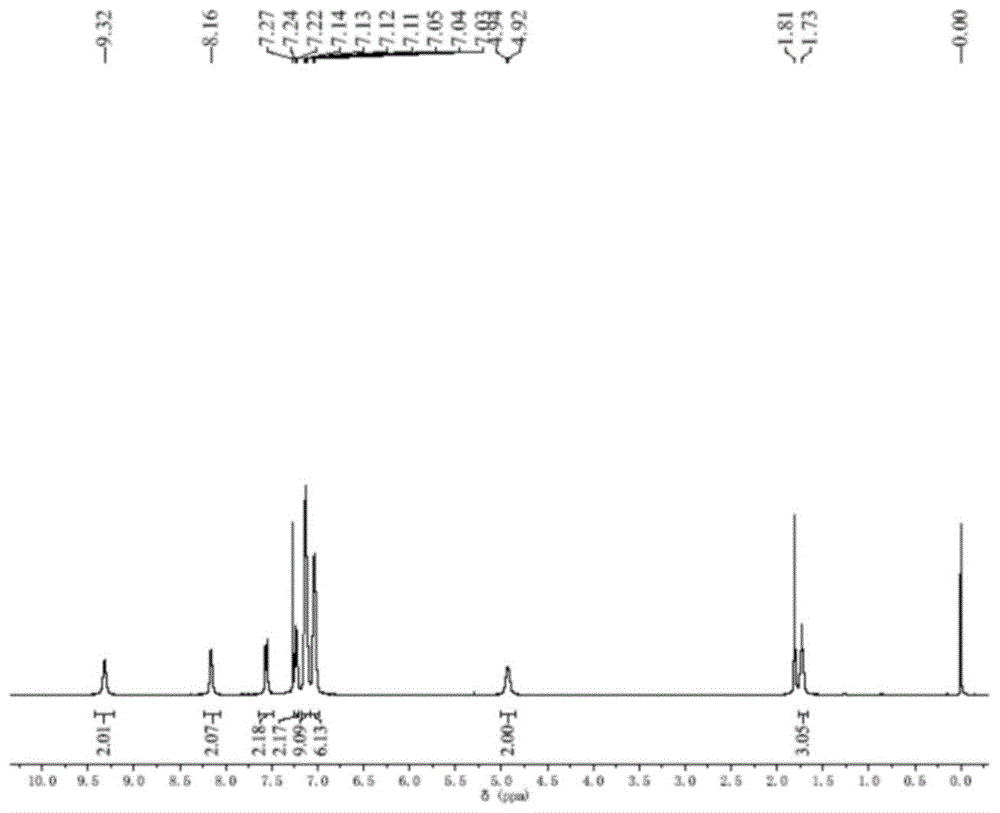
图1为tpepye在氘代氯仿中的核磁共振氢谱图;
图2为tpepye在氘代氯仿中的核磁共振碳谱图;
图3为tpepye的高分辨质谱图;
图4为实施例1中的荧光探针化合物的合成路径;
图5为实施例2中用于展示标准荧光曲线的荧光曲线图;
图6为实施例2中用于展示待测荧光曲线的荧光曲线图;
图7为实施例3中用于展示标准荧光曲线的荧光曲线图;
图8为实施例3中用于展示待测荧光曲线的荧光曲线图;
图9为实施例4中用于展示标准荧光曲线的荧光曲线图;
图10为实施例4中用于展示待测荧光曲线的荧光曲线图;
图11为实施例5中用于展示标准荧光曲线的荧光曲线图;
图12为实施例5中用于展示待测荧光曲线的荧光曲线图;
图13为实施例6中内毒素含量为0-10eu/ml的标准荧光曲线;
图14为实施例6中内毒素含量为0-0.25eu/ml的标准荧光曲线;
图15为实施例6中内毒素含量为0-10eu/ml的荧光强度曲线;
图16为实施例6中内毒素含量为0-2.5eu/ml的荧光强度曲线。
具体实施方式
以下结合附图及实施例,对本发明作进一步详细说明。
本发明中,无热原水为内毒素检测用水(内毒素含量<0.003eu/ml,视为0eu/ml);
本发明中所使用的注射液,包括0.9%氯化钠注射液、5%葡萄糖注射液、盐酸氨溴索注射液和注射用奥美拉唑钠,其初始内毒素含量均小于0.003eu/ml。因此,在实施过程中,我们可将所用注射液的初始内毒素含量视为0eu/ml。
本发明中,进行检测时均采用无热原移液枪头,所有玻璃瓶都在250℃的高温下烘烤半小时以上,以去除热源。
实施例1
一种荧光探针化合物,荧光探针化合物为(tpepye),分子式为c33h28ni,结构式为:
荧光探针化合物的制备方法,包括以下步骤:
s1.在250ml两口烧瓶中加入3.7g(22mmol)二苯基甲烷(1);对两口烧瓶抽真空后再充入氮气,重复三次;注入40ml无水四氢呋喃(thf);待原料溶解,将反应体系冷却到0℃,然后滴加13.75ml(22mmol)正丁基锂(1.6m),反应液变为红色;在0℃下搅拌1小时后(其他实施例可在1℃、2℃、3℃、4℃下搅拌1小时),将5.222g(20mmol)4-溴二苯甲酮(2)溶解在10mlthf中,然后缓慢滴加到反应体系中;将反应混合物自然升温至室温并继续反应10小时。反应结束后,用饱和氯化铵(nh4cl)溶液终止反应,粗产物用二氯甲烷(dcm)萃取三次。将有机相合并,分别用盐水和去离子水洗涤三次,然后用无水硫酸镁干燥,过滤,旋蒸,得到的固体用正己烷通过抽滤装置洗涤五次以除去过量的二苯基甲烷(1)。最终得到6.724g白色固体状的第一中间体(3),产率78.3%,将第一中间体(3)直接用于下一步脱水反应。
s2.在100ml圆底烧瓶中加入上述制备的第一中间体(3)和催化当量的对甲苯磺酸;在向体系中加入20ml甲苯后,升温至回流反应12小时。在反应液冷却到室温后,旋蒸除去甲苯,粗产物用硅胶色谱柱以石油醚为淋洗液进行分离,得到5.585g白色固体状的第二中间体(4),其总产率为67.9%。
s3.在氮气氛围下,将第二中间体(4)与4-硼酸吡啶溶解在甲苯中,加入碳酸钾水溶液(2m)和10mg四丁基溴化铵(tbab),并在室温下搅拌30分钟。然后加入四(三苯基磷)钯(pd(pph3)4)催化剂,并加热至80℃,反应16小时;反应结束后,待反应物冷却到室温时,用饱和食盐水洗涤,再用乙酸乙酯萃取三次;收集有机相,分别用盐水和去离子水洗涤多次,然后用无水硫酸镁干燥,过滤,旋蒸。粗产物用硅胶色谱柱以二氯甲烷/石油醚(v:v,1:3)为淋洗液进行分离,最终得到白色固体状的第三中间体(5)。
s4.将第三中间体(5)溶解于dmf中,加入碘乙烷,在80℃条件下搅拌反应12小时。粗产物通过重结晶提纯,最终得到黄色固体状的4-(4-(1,2,2-三苯基乙烯基)苯基)乙基吡啶碘盐(tpepye)。
tpepye在氘代氯仿中的核磁共振氢谱图如图1所示;
tpepye在氘代氯仿中的核磁共振碳谱图如图2所示;
tpepye的高分辨质谱图如图3所示;
tpepye合成路线如图4所示。
实施例2
本实施例中,待测溶液为:浓度为0.9%的氯化钠注射液内毒素含量为0.75eu/ml的阳性待测样品以及浓度为0.9%的氯化钠注射液内毒素含量为0.1eu/ml的阴性待测样品。
一种利用实施例1中的荧光探针化合物检测浓度为0.9%的氯化钠注射液中内毒素含量的方法,包括以下步骤:
(1)将荧光探针化合物溶解于无热原水中,制备储存液,具体的:取1.1mg荧光探针化合物tpepye,倒入25ml容量瓶中,在容量瓶中加入无热原水以定容,在涡旋震荡仪上震荡30s,使得tpepye充分溶解,制得浓度为100μm的tpepye溶液,即为储存液。
(2)标准品溶液配制:
取10eu内毒素工作标准品,加入10ml浓度为0.9%的氯化钠注射液中,配置成内毒素含量为1eu/ml的浓度为0.9%的氯化钠注射液标准品溶液;
将内毒素含量为1eu/ml的浓度为0.9%的氯化钠注射液标准品溶液通过浓度为0.9%的氯化钠注射液逐级稀释,得到内毒素含量依次为0.5eu/ml、0.25eu/ml的浓度为0.9%的氯化钠注射液标准品溶液;
取10ml未加入内毒素工作标准品的浓度为0.9%的氯化钠注射液作为阴性对照样品,其内毒素含量为0eu/ml。
(3)取储存液与标准品溶液混合均匀得标准反应液,取储存液与待测溶液混合均匀得待测反应液;
标准反应液配制方法如下:
依次取内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml的标准品溶液以及阴性对照样品各3ml,依次分别加入0.3ml的存储液,通过涡旋震荡器混合5s以混合均匀后依次形成内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml、和0eu/ml的标准反应液。
待测反应液配制方法如下:
分别取3ml浓度为0.9%的氯化钠注射液内毒素含量为0.75eu/ml的阳性待测样品以及浓度为0.9%的氯化钠注射液内毒素含量为0.1eu/ml的阴性待测样品,分别加入0.3ml存储液,通过涡旋震荡器震荡以均匀混合,形成阳性待测反应液以及阴性待测反应液。
(4)检测标准反应液得标准荧光曲线,检测待测反应液得待测荧光曲线;
将各种内毒素含量的标准反应液放入石英比色皿中,通过荧光分光光度计分别检测得出内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml和0eu/ml的标准荧光曲线,如图5所示。
将待测反应液放入石英比色皿中,通过荧光分光光度计检测得出阳性待测样品以及阴性待测样品的待测荧光曲线,如图6所示。
(5)对比待测荧光曲线与标准荧光曲线得出待测溶液中的内毒素含量是否达标;
《中国药典》中规定氯化钠注射液的内毒素含量应不高于0.5eu/ml。
如图6所示,图中的阳性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线上方,显示为阳性;
如图6所示,图中的阴性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线下方,显示为阴性;
可见尽管氯化钠注射液具有可转换碘离子的特性,但使用tpepye作为荧光探针进行检测时,不会出现假阳性的情况。
实施例3
本实施例中,待测溶液为:浓度为5%的葡萄糖注射液内毒素含量为0.75eu/ml的阳性待测样品以及浓度为5%的葡萄糖注射液内毒素含量为0.1eu/ml的阴性待测样品。
一种利用实施例1中的荧光探针化合物检测浓度为5%的葡萄糖注射液中内毒素含量的方法,包括以下步骤:
(1)将荧光探针化合物溶解于无热原水中,制备储存液,具体的:取1.65mg荧光探针化合物tpepye,倒入25ml容量瓶中,在容量瓶中加入无热原水以定容,在涡旋震荡仪上震荡30s,使得tpepye充分溶解,制得浓度为150μm的tpepye溶液,即为储存液。
(2)标准品溶液配制:
取10eu内毒素工作标准品,加入10ml浓度为5%的葡萄糖注射液中,配置成内毒素含量为1eu/ml的浓度为5%的葡萄糖注射液标准品溶液;
将内毒素含量为1eu/ml的浓度为5%的葡萄糖注射液标准品溶液用浓度为5%的葡萄糖注射液标准品溶液逐级稀释,得到内毒素含量依次为0.5eu/ml和0.25eu/ml的浓度为5%的葡萄糖注射液标准品溶液;
取10ml未加入内毒素工作标准品的浓度为5%的葡萄糖注射液作为阴性对照样品,其内毒素含量为0eu/ml。
(3)取储存液与标准品溶液混合均匀得标准反应液,取储存液与待测溶液混合均匀得待测反应液;
标准反应液配制方法如下:
依次取内毒素含量为1eu/ml、0.5eu/ml和0.25eu/ml的标准品溶液以及阴性对照样品各3ml,依次分别加入0.3ml的存储液,通过涡旋震荡器混合5s以混合均匀后依次形成内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml和0eu/ml的标准反应液。
待测反应液配制方法如下:
分别取3ml浓度为5%的葡萄糖注射液内毒素含量为0.75eu/ml的阳性待测样品以及浓度为5%的葡萄糖注射液内毒素含量为0.1eu/ml的阴性待测样品,分别加入0.3ml存储液,通过涡旋震荡器震荡以均匀混合,形成阳性待测反应液以及阴性待测反应液。
(4)检测标准反应液得标准荧光曲线,检测待测反应液得待测荧光曲线;
将各种内毒素含量的标准反应液放入石英比色皿中,通过荧光分光光度计分别检测得出内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml和0eu/ml的标准荧光曲线,如图7所示。
将待测反应液放入石英比色皿中,通过荧光分光光度计检测得出阳性待测样品以及阴性待测样品的待测荧光曲线,如图8所示。
(5)对比待测荧光曲线与标准荧光曲线得出待测溶液中的内毒素含量是否达标;
《中国药典》中规定葡萄糖注射液的内毒素含量应不高于0.5eu/ml。
如图8所示,图中的阳性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线上方,显示为阳性;
如图8所示,图中的阴性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线下方,显示为阴性。
实施例4
本实施例中,待测溶液为:浓度为0.25mg/ml的注射用奥美拉唑钠溶液内毒素含量为0.833eu/ml的阳性待测样品以及浓度为0.25mg/ml的注射用奥美拉唑钠溶液内毒素含量为0.1eu/ml的阴性待测样品。
一种利用实施例1中的荧光探针化合物检测注射用奥美拉唑钠中内毒素含量的方法,包括以下步骤:
(1)将荧光探针化合物溶解于无热原水中,制备储存液,具体的:取2.2mg荧光探针化合物tpepye,倒入25ml容量瓶中,在容量瓶中加入无热原水以定容,在涡旋震荡仪上震荡30s,使得tpepye充分溶解,制得浓度为200μm的tpepye溶液,即为储存液。
(2)标准品溶液配制:
将注射用奥美拉唑钠溶解于无热原水中,制备成溶液,具体的:取一瓶奥美拉唑钠(40mg),加入无热原用水溶解,定容到160ml,得到浓度为0.25mg/ml的注射用奥美拉唑钠溶液。
取10eu内毒素工作标准品,加入10ml注射用奥美拉唑钠溶液(0.25mg/ml)中,配置成内毒素含量为1eu/ml的注射用奥美拉唑钠标准品溶液;
将内毒素含量为1eu/ml的注射用奥美拉唑钠标准品溶液用0.25mg/ml的注射用奥美拉唑钠溶液逐级稀释,得到内毒素含量依次为0.5eu/ml和0.25eu/ml的注射用奥美拉唑钠标准品溶液;
取10ml未加入内毒素工作标准品的注射用奥美拉唑钠溶液(0.25mg/ml)作为阴性对照样品,其内毒素含量为0eu/ml。
(3)取储存液与标准品溶液混合均匀得标准反应液,取储存液与待测溶液混合均匀得待测反应液;
标准反应液配制方法如下:
依次取内毒素含量为1eu/ml、0.5eu/ml和0.25eu/ml的标准品溶液以及阴性对照样品各3ml,依次分别加入0.3ml的存储液,通过涡旋震荡器混合5s以混合均匀后依次形成内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml和0eu/ml的标准反应液。
待测反应液配制方法如下:
将注射用奥美拉唑钠待测样品以及内毒素标准品溶解于无热原用水中,配制成浓度为0.25mg/ml内毒素含量分别为0.833eu/ml、0.1eu/ml的溶液,分别取3ml浓度为0.25mg/ml的注射用奥美拉唑钠溶液内毒素含量为0.833eu/ml的阳性待测样品以及浓度为0.25mg/ml的注射用奥美拉唑钠溶液内毒素含量为0.1eu/ml的阴性待测样品,分别加入0.3ml存储液,通过涡旋震荡器震荡以均匀混合,形成阳性待测反应液以及阴性待测反应液。
(4)检测标准反应液得标准荧光曲线,检测待测反应液得待测荧光曲线;
将各种内毒素含量的标准反应液放入石英比色皿中,通过荧光分光光度计分别检测得出内毒素含量为1eu/ml、0.5eu/ml、0.25eu/ml、和0eu/ml的标准荧光曲线,如图9所示。
将待测反应液放入石英比色皿中,通过荧光分光光度计检测得出阳性待测样品以及阴性待测样品的待测荧光曲线,如图10所示。
(5)对比待测荧光曲线与标准荧光曲线得出待测溶液中的内毒素含量是否达标;
《中国药典》中规定注射用奥美拉唑钠的内毒素含量应不高于2eu/mg,本实施例中将注射用奥美拉唑钠配制成浓度为0.25mg/ml的溶液,再进行检测,所以溶液样品的内毒素限值为0.5eu/ml。
如图10所示,图中的阳性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线上方,显示为阳性;
如图10所示,图中的阴性待测反应液所测得的荧光曲线位于0.5eu/ml的标准荧光曲线下方,显示为阴性。
实施例5
本实施例中,待测溶液为:浓度为0.8mg/ml的盐酸氨溴索注射液稀溶液内毒素含量为0.625eu/ml的阳性待测样品以及浓度为0.8mg/ml的盐酸氨溴索注射液稀溶液内毒素含量为0.1eu/ml的阴性待测样品。
一种利用实施例1中的荧光探针化合物检测盐酸氨溴索注射液(2ml包装,每支含15mg盐酸氨溴索)中内毒素含量的方法,包括以下步骤:
(1)将荧光探针化合物溶解于无热原水中,制备储存液,具体的:取1.1mg荧光探针化合物tpepye,倒入25ml容量瓶中,在容量瓶中加入无热原水以定容,在涡旋震荡仪上震荡30s,使得tpepye充分溶解,制得浓度为100μm的tpepye溶液,即为储存液。
(2)标准品溶液配制:
将盐酸氨溴索注射液(2ml包装,每支含15mg盐酸氨溴索)用无热原用水进行稀释,制备成稀溶液,具体的:取1支盐酸氨溴索注射液,加入无热原用水稀释,定容到18.75ml,得到浓度为0.8mg/ml的盐酸氨溴索注射液稀溶液。
取8eu内毒素工作标准品,加入10ml盐酸氨溴索注射液稀溶液(0.8mg/ml)中,配置成内毒素含量为0.8eu/ml的盐酸氨溴索注射液标准品溶液;
将内毒素含量为0.8eu/ml的盐酸氨溴索注射液标准品溶液用0.8mg/ml的盐酸氨溴索注射液稀溶液逐级稀释,得到内毒素含量依次为0.4eu/ml和0.2eu/ml的盐酸氨溴索注射液标准品溶液;
取10ml未加入内毒素工作标准品的盐酸氨溴索注射液稀溶液(0.8mg/ml)作为阴性对照样品,其内毒素含量为0eu/ml。
(3)取储存液与标准品溶液混合均匀得标准反应液,取储存液与待测溶液混合均匀得待测反应液;
标准反应液配制方法如下:
依次取内毒素含量为0.8eu/ml、0.4eu/ml、和0.2eu/ml的标准品溶液以及阴性对照样品各3ml,依次分别加入0.3ml的存储液,通过涡旋震荡器混合5s以混合均匀后依次形成内毒素含量为0.8eu/ml、0.4eu/ml、0.2eu/ml、和0eu/ml的标准反应液。
待测反应液配制方法如下:
将盐酸氨溴索注射液(2ml包装,每支含15mg盐酸氨溴索)待测样品以及内毒素标准品用无热原用水稀释,配制成浓度为0.8mg/ml内毒素含量分别为0.625eu/ml、0.1eu/ml的稀溶液,分别取3ml浓度为0.8mg/ml的盐酸氨溴索注射液稀溶液内毒素含量为0.625eu/ml的阳性待测样品以及浓度为0.8mg/ml的盐酸氨溴索注射液稀溶液内毒素含量为0.1eu/ml的阴性待测样品,分别加入0.3ml存储液,通过涡旋震荡器震荡以均匀混合,形成阳性待测反应液以及阴性待测反应液。
(4)检测标准反应液得标准荧光曲线,检测待测反应液得待测荧光曲线;
将各种内毒素含量的标准反应液放入石英比色皿中,通过荧光分光光度计分别检测得出内毒素含量为0.8eu/ml、0.4eu/ml、0.2eu/ml和0eu/ml的标准荧光曲线,如图11所示。
将待测反应液放入石英比色皿中,通过荧光分光光度计检测得出阳性待测样品以及阴性待测样品的待测荧光曲线,如图12所示。
(5)对比待测荧光曲线与标准荧光曲线得出待测溶液中的内毒素含量是否达标;
《中国药典》中规定盐酸氨溴索注射液的内毒素含量应不高于0.5eu/mg,本实施例中将盐酸氨溴索注射液稀释成浓度为0.8mg/ml的稀溶液,再进行检测,所以溶液样品的内毒素限值为0.4eu/ml。
如图12所示,图中的阳性待测反应液所测得的荧光曲线位于0.4eu/ml的标准荧光曲线上方,显示为阳性;
如图12所示,图中的阴性待测反应液所测得的荧光曲线位于0.4eu/ml的标准荧光曲线下方,显示为阴性;
可见尽管盐酸氨溴索注射液具有可转换碘离子的特性,但使用tpepye作为荧光探针进行检测时,不会出现假阳性的情况。
实施例6
一种荧光探针化合物检测范围的测定方法,包括以下步骤:
a.将荧光探针化合物溶解于无热原水中,制备储存液,浓度为100μm;
b.将一定量的内毒素标准品溶解于无热原水中,制得内毒素含量分别为0eu/ml、0.005eu/ml、0.01eu/ml、0.025eu/ml、0.05eu/ml、0.1eu/ml、0.25eu/ml、0.5eu/ml、1eu/ml、2.5eu/ml、5eu/m、10eu/ml的标准溶液;
c.取0.3ml储存液与3ml标准溶液混合均匀得标准反应液;
d.用荧光分光光度计,检测标准反应液得标准荧光曲线以及荧光强度曲线。
标准荧光曲线见图13-14,荧光强度曲线见图15-16。
图15是在540nm处,含有内毒素的标准反应液(0.005-10eu/ml)的荧光强度与不含内毒素的标准反应液(0eu/ml)的荧光强度的比值i/i0,其与内毒素含量的关系;
图16是在0-2.5eu/ml的浓度范围内,标准反应液在540nm处的荧光强度与其内毒素含量的线性关系。
根据图13和图14可得,在0.05-10eu/ml的浓度范围内,随着内毒素含量的提高,荧光曲线逐渐升高;在0-0.025eu/ml的浓度范围内,随着内毒素含量的提高,荧光曲线几乎无变化,因此可见,在该检测条件下,tpepye的检测极限为0.05eu/ml。
根据图15和图16可得,在0-10eu/ml的浓度范围内,随着内毒素含量的提高,荧光曲线在540nm处的强度相应提高;尤其在0.05-2.5eu/ml的浓度范围内,随着内毒素含量的提高,荧光曲线在540nm处的强度线性增加且线性相关系数高达0.999,因此可见,tpepye对内毒素的粗检测范围能包含0.05-10eu/ml足以满足大部分《中国药典》中规定的上限值的检测,且tpepye对内毒素的精确检测范围在0.05-2.5eu/ml。
本具体实施方式的实施例均为本发明的较佳实施例,并非依此限制本发明的保护范围,故:凡依本发明的结构、形状、原理所做的等效变化,均应涵盖于本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!