一种利用枯草芽孢杆菌表达系统快速、高效筛选抗菌基因的方法与流程
本发明属于分子生物学领域,涉及一种利用枯草芽孢杆菌表达系统快速、高效筛选抗性基因的方法,它主要用于快速找到抗性基因,并能验证基因功能。
背景技术:
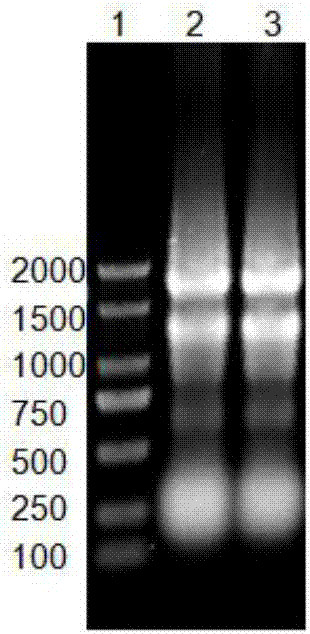
:枯草芽孢杆菌是经美国fda认证的一种对环境无害的(gras)革兰氏阳性细菌,全基因组测序已经完成,其作为工程菌具有很大的优势,原因在于:芽孢杆菌的胞外蛋白可以直接分泌到培养基中;芽孢杆菌分泌的蛋白无内毒素,较为安全;被认为非常适合遗传操作,并已成为被广泛应用于实验室研究的模式生物,本研究中所用芽孢杆菌wb800,已经突变掉了8个主要的胞外蛋白酶,大大降低了抗菌肽降解的可能性。cdna文库是以特定的组织或细胞的mrna为模板,逆转录形成的cdna,再利用体外的dna重组技术,转入合适的受体进行扩增或表达,形成一个重组的克隆群体,cdna文库是目前研究基因功能最常用的手段之一。抗菌肽(antimicrobialpeptide,amp)是由生物合成的具有广谱抗性的小分子多肽,到目前为止已有1200多种来自动植物的抗菌肽被陆续发现,其中70多种抗菌多肽的结构已经被揭示,抗菌肽对多种真菌、细菌、寄生虫和病毒都有一定的杀伤作用引起了广泛的重视。目前抗菌肽的相关研究大多集中在活性物质的提取分离和纯化方面以及抗性机理的研究上,分离抗菌肽的方法主要有三种:1、生物体直接提取纯化,但是抗菌肽天然合成量非常少,分离纯化过程中损失大,且容易失活;2、人工合成已知的抗菌肽,此方法成本高、耗时长,无法保证抗菌肽的天然构象和生物活性,而且难以筛选到新的抗菌肽;3、构建工程菌融合表达抗菌基因,这种方法利用差异显示技术,通过pcr的手段将有差异的片段分开,筛选出目的基因,经pcr扩增后,片段的重复率显著增加,且分离的片段可能为无效片段,此外,另一种方法是基于蛋白或多肽的逐级分离检测,从活性蛋白入手推断出的基因序列再合成抗菌肽,此方法工作周期长,工作量大,且再合成的抗菌肽往往没有预期的活性。本发明采用芽孢杆菌wb800分泌系统,首先抗菌肽在菌体内部表达,使得筛选系统变得更灵敏,其次,采用文库筛选的方法可以在是筛选出很多个潜在的抗菌肽的同时,对应上相应基因片段,最后,芽孢杆菌本身就是一个生防菌,发现的新的抗菌肽具有很大的应用潜力。技术实现要素:本发明的目的在于提供一种利用枯草芽孢杆菌表达系统快速、高效筛选抗菌基因的方法,本方法首先诱导植物中抗性基因的表达,再选择合适的载体构建一个完整的cdna文库,导入到枯草芽孢杆菌后,利用其分泌表达系统表达重组质粒,点样法筛选抗性表型,实验步骤简单、易行,适合工作量大的文库筛选,能够在有限的时间内快速高效的得到具有抗性表型的克隆,为筛选抗性基因提了新的途径。为了实现上述的目的,本发明采用以下技术措施:一种利用枯草芽孢杆菌表达系统快速、高效筛选抗菌基因的方法,其步骤为:1)含目标基因的cdna文库的制备:a.在健康植株上接种病原菌(该病原菌必须可以在寄主植物上致病,对病原菌种类无要求)诱导抗性基因的表达后,提取目标植物总rna;b.分离纯化mrna;c.设计含xbaⅰ酶切位点的oligodt,其序列为5′-acaggctctagagcttttttttttttttttttttttt-3′,合成cdna第一条链,以第一条链为模板合成第二条cdna链;cdna合成完成后,加上含ndeⅰ酶切位点的的接头,所述的含ndeⅰ酶切位点的的接头有3对,以减少移码突变的概率,由下述方法制得:将ndeⅰ1和ndeⅰ2,ndeⅰ3和ndeⅰ4,ndeⅰ5和ndeⅰ6等量混合,并加入1/10体积的10×pcrbuffer,在pcr仪中95℃加热5分钟,然后自然冷却至室温,形成接头ndeⅰ1/ndeⅰ2,ndeⅰ3/ndeⅰ4,ndeⅰ5/ndeⅰ6;将接头ndeⅰ1/ndeⅰ2,ndeⅰ3/ndeⅰ4和ndeⅰ5/ndeⅰ6等量混合,保存于-20℃,备用。d.cdna连接载体:将步骤c中的加完接头的cdna用限制性核酸内切酶xbaⅰ和ndeⅰ双酶切后,连接到质粒pbe-s上;e.将连接体系转化到大肠杆菌高效感受态细胞中,扩增质粒;群体提质粒后转化枯草芽孢杆菌wb800,挑取不少于2000个转化子,保存备用;2)致死菌株的筛选a.将转化子在标记卡那霉素抗性的lb培养基中摇培过夜后,于相同抗性的平板上点样,筛选出原本长势正常但后期变成透明或半透明状的菌株,初步确认,该基因具有致死作用;b.将上述初步确认的含致死基因菌液摇培后提取质粒,经大肠杆菌扩增后重新转化枯草芽孢杆菌wb800和枯草芽孢杆菌野生菌株168,均出现了致死表型,表明致死表型由插入的基因引起,该基因作为下一步的候选基因;3)抗线虫基因的筛选采用滤纸片法测定含候选基因的转化子对秀丽隐杆线虫的抗性,以枯草芽孢杆菌wb800作为对照,测定选择指数,选择指数公式如下:选择指数=(测试菌中线虫数量-对照菌中线虫数量)/线虫总数,选择指数小于-30%的转化子具有抗线虫作用,用于下一步的筛选;4)抗菌基因的筛选硫酸铵沉淀法提取步骤3)中含抗线虫基因的菌株的胞外粗蛋白,以空载体菌株为对照,滤纸片法测定粗蛋白的抑菌效果,抽提质粒和基因测序,由此得到相应的抗菌基因。优选地,步骤1)中将连接体系转化到大肠杆菌高效感受态细胞中,所述的感受态细胞为大肠杆菌感受态hst08,提高转化效率,减少基因的丢失。与现有技术相比,本发明具有以下优点及有益效果:本发明中致死筛选过程首先让抗菌肽在菌体内部表达,最后分泌到胞外,体内抗菌肽积累并起作用,使得筛选系统变得更加灵敏,且可以筛选出很多个潜在的抗菌肽的同时,对应上相应基因片段,抗线虫基因筛选和粗蛋白抑菌筛选,可以在众多的潜在抗菌肽中,筛选出具有抗线虫或抗菌作用的基因。最后,芽孢杆菌本身就是一个生防菌,发现的新的抗菌肽在直接应用方面具有很大的潜力。发现抗菌肽后,可以先通过基因测序确定基因序列,再通过各大数据库的比对来初步确定是否为新的抗菌基因,如果比对无结果,则可以进一步比对抗菌肽序列,最终确定是否为新的抗菌肽。附图说明图1为提取的总rna凝胶电泳检测图。共有3条泳道,1泳道为dl2000dnamaker,2、3泳道为总rna凝胶检测结果。18srrna、28srrna条带清晰。图2为纯化后的mrna凝胶电泳检测图。共有2条泳道,1泳道为dl2000dnamaker,2泳道为mrna凝胶检测结果。mrna为弥散状条带。图3为双链cdna凝胶电泳检测图。共有2条泳道,1泳道为100bpdnaladder,2泳道为cdna凝胶检测结果。cdna为弥散状条带。图4、图5分别为板蓝根cdna文库和半夏cdna文库插入片段随机pcr检测图。共50条泳道,13泳道为100bpdnaladder,其他泳道为随机挑选的文库中的菌株。图6为含抗性基因致死模式图。图a为点样12h后菌落形态图,图b为点样24h后菌落形态图。培养皿中左侧菌落为空载体菌落,即对照菌落,右侧菌落为致死菌株菌落,即测试菌菌落。图7为致死菌扫描电镜图片。图a为野生型枯草芽孢杆菌wb800菌株扫描电镜图,图b为转入pbe-s载体的空载体菌株扫描电镜图,图c为致死菌菌株扫描电镜图。图8为测试菌粗蛋白抑菌效果图。图中指示菌为野生型枯草芽孢杆菌wb800,白色圆圈为滤纸片,滤纸片周围出现抑菌圈的粗蛋白为测试菌粗蛋白,滤纸片周围没有出现抑菌圈的粗蛋白为空载体菌株粗蛋白。图9为滤纸片法筛选抗线虫基因效果图。图a为对照野生型枯草芽孢杆菌wb800滤纸片下线虫和虫道分布图,图b为测试菌滤纸片下线虫和虫道分布图。具体实施方式实施例1:板蓝根中抗菌基因的筛选1.板蓝根cdna文库的制备:(1)将板蓝根培育至4周后,接种玉米纹枯病病原菌agi-ia,封口膜密封保湿。诱导板蓝根抗性基因的大量表达,每隔6h取样一次,取完后立即于-70℃超低温保存备用,直至板蓝根叶片发病,不再取样。用细胞总rna提取试剂trizol,大量提取板蓝根叶片总rna,富集后浓度为2600ng/μl。取5μlrna凝胶电泳,检测rna质量(见图1),剩下的于-70℃超低温保存备用,直到总体积达到500μl,进行纯化。(2)磁珠法分离纯化mrna(isolationsystems),按照实验手册进行实验,如果纯化浓度低,可用多个磁珠富集,纯化出mrna后所测浓度为260ng/μl,取5μlmrna凝胶电泳,检测mrna质量(见图2),剩下mrna立即反转录。(3)设计含xbaⅰ酶切位点的oligodt合成cdna第一条链,以第一条链为模板合成cdna第二条链。cdna第一条链合成体系:mrna(5μg)11μl5×1ststrandsynthesisbuffer4μldntpmixture1μlrnaseinhibitor1μloligodtprimer(含xbaⅰ酶切位点)2μlrtnase1μl总体积20μl含xbaⅰ酶切位点的oligodt的序列为5’-acaggctctagagcttttttttttttttttttttttt-3’,用于mrna纯化过程中结合mrna。mrna65℃加热5min后,立即取出冰中放置5min(冷却)。加入上述反应液室温放置10min,转移到42℃恒温水浴槽内(可以在pcr仪中做)反应1h。反应结束后放置冰中冷却2min。cdna第二条链合成体系:在第一条链合成后的微量离心管中再加入下面反应液,全量142μl:5×2ststrandsynthesisbuffer30μldntpmixture3μlrnase-freeh2o89μl加入2μle.colidnapolymerasel和2μle.colirnaseh/e.colidnaligasemixture,轻轻混匀,16℃反应2h;70℃加热10min,取出室温放置5min;加入4μl的t4dnapolymerase,轻轻混匀,37℃反应10min;向第二条链cdna合成反应液(150μl)中加入rnase-freeh2o至终体积500μl,再加入等量的苯酚/氯仿/异戊醇(25:24:1)溶液,vortex震荡5-10s;室温下15000rpm离心5min,小心取出上层水相至另一个新的微量离心管中;加入1/10体积(50μl)的3m醋酸钠(ph5.2),2.5倍量的100%乙醇,均匀混合后,立即在15000rpm下离心30min;弃上清,用70%乙醇清洗,可重复一次;干燥沉淀后,用20μl的rnase-freeh2o溶解沉淀(检测结果见图3),浓度为1300ng/μl,于-20℃保存,便于后面加接头用。制备含ndeⅰ酶切位点的接头的方法:按照合成单上的说明将合成的寡核苷酸溶于相应体积的双蒸水或tebuffer中,使浓度为100pm/μl。在pcr管中分别将ndeⅰ1和ndeⅰ2,ndeⅰ3和ndeⅰ4,ndeⅰ5和ndeⅰ6等量混合,并加入1/10体积的10×pcrbuffer。在pcr仪中95℃加热5分钟,然后自然冷却至室温(20-25℃,以下相同)。形成的接头如下:再将接头ndeⅰ1/ndeⅰ2,ndeⅰ3/ndeⅰ4和ndeⅰ5/ndeⅰ6等量混合,保存于-20℃,以备后用。加上含ndeⅰ酶切位点的接头(primescripttmdoublestrandcdnasynthesiskit),加ndeⅰ接头体系:cdna3μl10×t4dnaligasebuffer2μlndeⅰadaptor0.5μlt4dnaligase1μlddh2o13.5μl总体积20μl轻轻混匀后,16℃反应10h左右。70℃保温30min,室温放置5min后,立即双酶切处理。双酶切载体体系:轻轻混匀后,37℃反应3-4h。利用pcr切胶回收试剂盒回收双酶切产物中的大片段,用15μl-20μl的ddh2o洗脱,用分光光度计检测浓度为1000ng/μl,回收产物转化大肠杆菌检测是否酶切完全。双酶切cdna体系:10×mbuffer4μlndeⅰ2μlxbaⅰ2μlcdna20μlddh2o12μl总体积20μl轻轻混匀后,37℃反应3-4h。利用pcr清洁回收试剂盒去除双酶切产物,用15μl-20μl的ddh2o洗脱,用分光光度计检测浓度为1000ng/μl,以备后面连接载体。(4)cdna连接载体:将加完接头的cdna双酶切后连到含173种信号肽的信号肽库质粒pbe-s(购自宝生物工程有限公司)上,重组蛋白的表达分泌和信号肽的种类密切相关,在载体上连接一个信号肽库,有利于筛选不同的分泌蛋白。连接体系:cdna12μlpbe-s1μl10xt4dnaligasebuffer2μlt4dnaligase1μlddh2o4μl总体积20μlpcr仪中16℃保温3-5h,此20μl连接溶液可以做后面转化用。(5)将连接体系先转化到大肠杆菌高效感受态hst08(购自武汉友名生物工程有限公司)中,涂平板大量扩增重组质粒,群体提质粒后再转化枯草芽孢杆菌wb800菌株(购自上海北诺生物科技有限公司),挑取挑取不少于2000个转化子,保存备用,以便进行后续重组蛋白的分泌表达。2.致死菌株的筛选(1)将转化子在标记抗性卡那霉素终浓度为10μg/ml的lb培养基中摇培过夜后,于相同抗性的平板上点样,定期观察菌落形态,发现原本长势很好的菌株,在生长到一定时间后开始慢慢死去,菌落从厚实致密的形态慢慢消解,变成透明或半透明状(见图6),扫描电镜观察致死菌株菌落形态,发现该菌株菌体扭曲形变或中间变空(见图7)。初步确认,该基因具有致死作用。我们用这个方法共筛选到60个致死菌株,选取编号为915的菌株进行后续验证。(2)将915的菌株摇培8-12h后提取质粒,重新转化枯草芽孢杆菌wb800,仍出现致死表型,将该质粒转化枯草芽孢杆菌野生菌株168(由本实验室保存)中,也出现了致死表型,而空载体菌株不出现致死现象。进一步表明致死表型由插入的基因引起,即插入的基因起致死作用,该基因作为下一步的候选基因。3.抗线虫基因的筛选滤纸片法:于ngm平板两侧放上0.22μm的有机滤膜,将摇培72h后的915菌株与枯草芽孢杆菌wb800菌株分别置于滤膜上,生长12h后,揭开滤膜点上大肠杆菌op50,培养12h后,在中间点上l4stage秀丽隐杆线虫70-80只,10h后统计两侧菌落内的线虫数量(见图9),经多次、大量筛选后,得出的数据如下:915分泌物处共有线虫263只,另一侧wb800菌株分泌物处共有682只,以选择指数为指标(选择指数=(测试菌中线虫数量-对照菌中线虫数量)/线虫总数),915选择指数为-44.34%(选择指数小于-30%的转化子具有抗线虫作用),可以确定915菌株分泌物具有一定的抗线虫作用,用于下一步的筛选。4.抗菌基因的筛选将915菌株在含卡那霉素终浓度为10μg/ml的抗性lb平板上活化2代之后转入50ml相同抗性lb中摇培12h,再按2%接种量接种至200ml相同抗性lb中于37℃中摇培发酵72h,取30ml上清用硫酸铵沉淀的方法提取粗蛋白,用pbs溶解蛋白,透析除去盐离子。以空载体菌株为对照,滤纸片法测定该基因的抑菌效果,发现915菌株对枯草芽孢杆菌wb800(见图8)、空载体菌株、青枯病原菌、白叶枯病原菌等多种细菌具有一定的抑菌效果。对915菌株进行质粒提取,并测序拿到基因序列后,在各大数据库中并未比对到任何结果,将对应的蛋白序列进行比对,也没有比对到结果,可以确定,该基因为新的抗菌基因。实施例2:半夏中抗菌基因的筛选1.半夏cdna文库的制备:将健康的半夏从培养钵中取出,块茎用无菌水冲洗干净后,用无菌的注射器针头在每个健康的块茎上扎7-10个孔,放入摇培好的半夏软腐病菌菌液中浸泡15-20min后取出,重新栽种到培养钵中,培养箱内培养,诱导半夏抗性基因的大量表达,每隔6h取样一次,取完后立即于-70℃超低温保存备用,直至半夏根基发病,不再取样。用细胞总rna提取试剂trizol,大量提取半夏叶片总rna,富集后浓度为9675ng/μl。取5μlrna进行凝胶电泳,检测rna质量,剩下的于-70℃超低温保存备用,直到总体积达到500μl,进行纯化。(2)磁珠法分离纯化mrna(isolationsystems),按照实验手册进行实验,如果纯化浓度低,可用多个磁珠富集,纯化出mrna后所测浓度为188ng/μl,取5μlmrna凝胶电泳,检测mrna质量,剩下mrna立即反转录。(3)设计含xbaⅰ酶切位点的oligodt合成cdna第一条链,以第一条链为模板合成cdna第二条链。cdna第一条链合成体系:含xbaⅰ酶切位点的oligodt的序列为5′-acaggctctagagcttttttttttttttttttttttt-3′,用于mrna纯化过程中结合mrna。mrna65℃加热5min后,立即取出冰中放置5min(冷却)。加入上述反应液室温放置10min,转移到42℃恒温水浴槽内(可以在pcr仪中做)反应1h。反应结束后放置冰中冷却2min。cdna第二条链合成体系:在第一条链合成后的微量离心管中再加入下面反应液,全量142μl:加入2μle.colidnapolymerasel和2μle.colirnaseh/e.colidnaligasemixture,轻轻混匀,16℃反应2h;70℃加热10min,取出室温放置5min;加入4μl的t4dnapolymerase,轻轻混匀,37℃反应10min;向第二条链cdna合成反应液(150μl)中加入rnase-freeh2o至终体积500μl,再加入等量的苯酚/氯仿/异戊醇(25:24:1)溶液,vortex震荡5-10s;室温下15000rpm离心5min,小心取出上层水相至另一个新的微量离心管中;加入1/10体积(50μl)的3m醋酸钠(ph5.2),2.5倍量的100%乙醇,均匀混合后,立即在15000rpm下离心30min;弃上清,用70%乙醇清洗,可重复一次;干燥沉淀后,用20μl的rnase-freeh2o溶解沉淀,浓度为567ng/μl,于-20℃保存,便于后面加接头用。含ndeⅰ酶切位点的接头的制备方法参见实施例1。加含ndeⅰ酶切位点的接头体系:cdna2μl10×t4dnaligasebuffer2μlndeⅰadaptor1μlt4dnaligase1μlddh2o14μl总体积20μl轻轻混匀后,16℃反应10h左右。65℃保温30min,室温放置5min后,立即双酶切处理。双酶切cdna体系:10×mbuffer4μlndeⅰ2μlxbaⅰ2μl加过接头cdna20μlddh2o12μl总体积20μl轻轻混匀后,37℃反应3-4h。利用pcr清洁回收试剂盒去除双酶切产物,用40μl的ddh2o洗脱,用分光光度计检测浓度为23.5ng/μl,以备后面连接载体。摇培活化含空载体的大肠杆菌菌株,按照试剂盒方法提取质粒,浓度为550ng/μl。双酶切载体体系:10×greenbuffer6μlndeⅰ3μlxbaⅰ3μlpbe-s7μlddh2o41μl总体积60μl轻轻混匀后,37℃反应3-4h。利用pcr切胶回收试剂盒回收双酶切产物中的大片段,用25μl的ddh2o洗脱,用分光光度计检测浓度为58ng/μl,回收产物转化大肠杆菌检测是否酶切完全。(4)cdna连接载体:将加完接头的cdna双酶切后连到含173种信号肽的信号肽库质粒pbe-s(购自大连宝生物科技有限公司)上,重组蛋白的表达分泌和信号肽的种类密切相关,在载体上连接一个信号肽库,有利于筛选不同的分泌蛋白。连接体系:cdna6.5μlpbe-s6μl10×t4dnaligasebuffer2μlt4dnaligase1μlddh2o4.5μl总体积20μlpcr仪中16℃保温3-5h,此20μl连接溶液可以-20℃保存,以便后面转化使用。(5)将连接体系先转化到大肠杆菌高效感受态hst08中,涂平板大量扩增重组质粒,群体提质粒,浓度为1958ng/μl。再转化枯草芽孢杆菌wb800菌株,挑取2039个转化子,保存备用,以便进行后续重组蛋白的分泌表达。2.致死菌株的筛选(1)将转化子在标记抗性kanamycin终浓度为10μg/ml的lb培养基中摇培过夜后,于相同抗性的平板上点样,定期观察菌落形态,发现原本长势很好的菌株,在生长到一定时间后开始慢慢死去,菌落从厚实致密的形态慢慢消解,变成透明或半透明状,扫描电镜观察致死菌株菌落形态,发现该菌株,菌体扭曲形变或中间变空。初步确认,该基因具有致死作用。我们用这个方法共筛选到95个致死菌株,选取编号为1366的菌株进行后续验证。(2)将1366菌株摇培8-12h后提取质粒,重新转化枯草芽孢杆菌wb800,仍出现致死表型,将该质粒转化枯草芽孢杆菌野生菌株168中,也出现了致死表型,而空载体菌株不出现致死现象。进一步表明致死表型由插入的基因引起,即插入的基因起致死作用,该基因作为下一步的候选基因。3.抗线虫基因的筛选滤纸片法:于ngm平板两侧放上0.22μm的有机滤膜,将摇培72h后的1366菌株与枯草芽孢杆菌wb800菌株分别置于滤膜上,生长12h后,揭开滤膜点上大肠杆菌op50,培养12h后,在中间点上l4stage秀丽隐杆线虫70-80只,10h后统计两侧菌落内的线虫数量,经多次、大量筛选后,得出的数据如下:1366分泌物处共有线虫644只,另一侧wb800菌株分泌物处共有1369只,以选择指数为指标(选择指数=(测试菌中线虫数量-对照菌中线虫数量)/线虫总数),1366选择指数为-36.02%(选择指数小于-30%的转化子具有抗线虫作用),可以确定1366菌株分泌物具有一定的抗线虫作用,用于下一步筛选。4.抗菌基因的筛选将1366菌株在含卡那霉素终浓度为10μg/ml的抗性lb平板上活化2代之后转入50ml相同抗性lb中摇培12h,在按2%接种量接种至200ml相同抗性lb中于37℃中摇培发酵72h,取30ml上清用硫酸铵沉淀的方法提取粗蛋白,用pbs溶解蛋白,透析除去盐离子。以空载体菌株为对照,滤纸片法测定该基因的抑菌效果,发现1366菌株对wb800、空载体菌株、青枯病原菌,白叶枯病原菌等多种细菌具有一定的抑菌效果。对1366菌株进行质粒提取,并测序拿到基因序列后,在各大数据库中并未比对到任何结果,将对应的蛋白序列进行比对,也没有比对到结果,可以确定,该基因为新的抗菌基因。以上列举的仅是本发明的2个具体实施例,本发明的实验材料可以是其他植物,例如大蒜、玉米等可食用作物,金银花、鱼腥草等药用植物,还可以是哈茨木霉等生防菌。抗菌基因筛选中选用的指示菌还可以是果斑病原菌、溃疡病原菌、水稻细菌性条斑病病原菌等各种死体营养性病原细菌。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1