针对目标核酸靶标的探针的制备方法与流程
本发明涉及分子生物学领域,具体而言,涉及一种针对目标核酸靶标的探针的制备方法。
背景技术:
荧光原位杂交(fluorescentinsituhybridization,fish)籍由杂交探针的本身的序列和荧光,能提供标记位点的在细胞核内的空间位置信息,一直以来与基于3c(chromatinconformationcapture,染色质构象捕获)的各种生物技术(如4c,5c,hic,chia-pet等)互补,成为研究染色质结构不可或缺的重要技术之一。传统的fish技术一般以含有目标物种来源的一段完整基因组片段(一般是bac,pac,yac等)作为模板,通过生物酶的作用进行片段化,之后进行荧光标记做出杂交探针,在固定细胞中,对特定的基因组片段,通过碱基互补配对原理,进行荧光标记并成像、获得具体的核内空间信息。但是传统的原位杂交技术受限于bac等模板本身的特性,具有准备时间长,所需模板量大,基因分辨率低(100-200kb),克隆中含有重复片段、需要加入物种特异的cot-1dna等缺点,在进行染色质结构研究中,对于大量存在的小于200kb的相互作用的标记适用性差,对于没有商业化cot-1dna的物种的研究更是捉襟见肘。因此,迫切需要开发具有快速高效、模板需求量低、基因组分辨率高、且不需要cot-1dna的荧光原位杂交方法,替代现有的传统fish技术方案。
目前来看,在已经报道的新型fish技术中,oligopaint技术,hd-fish技术,casfish技术和md-fish技术都针对上述4点进行了不同程度的优化,主要的进步在于提高的基因组分辨率(2.5kb-10kb)和无需加入cot-1dna来抑制重复序列。但是,这四种技术有些技术成本高昂,制备复杂,性价比低,有些技术需要生物信息学工具挖掘出合适的探针序列,对于一般的实验室而言很难直接运用。
有鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于提供一种针对目标核酸靶标的探针的制备方法,该方法包括:
a)获取感兴趣的靶dna序列;
b)使用转座酶,将所述靶dna序列进行片段化的同时,在片段化的dna序列两端加上接头序列;和
c)利用所述接头序列,获取所述片段化的dna序列,以产生探针。
根据本发明的再一方面,本发明还涉及进行杂交测定的方法,其包括利用如上所述的方法产生探针,并将靶核酸与所述探针接触。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
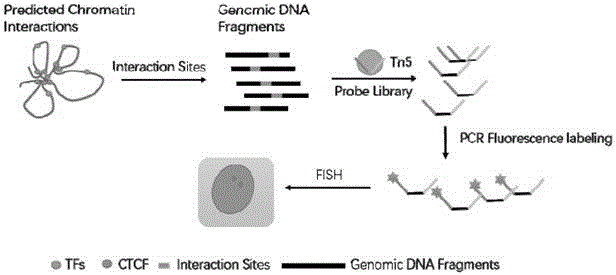
图1为本发明的一个实施方式的流程示意图;
图2为本发明的一个实施方式中,tn5-fish与传统bacfish联用在wtmesc细胞和platr22-komesc细胞中验证tn5-fish基因组位点的标记特异性的比较;
bac探针(绿色)和tn5-platr22探针(红色,图2a,2b)或tn5-gm19705探针(红色,图2c,2d)在wtmesc细胞(图2a,2c)或platr22-komesc细胞(图2b,2d)中同时进行杂交;
图3为本发明的一个实施方式中,k562细胞中tn5-fish和bacfish联用,验证100kb长度的染色质相互作用及kb基因组分辨率比较;
图4为本发明的一个实施方式中,多色tn5-fish对gm12878细胞中预测的位于chr2:227672028-227743852处两端具有相互作用位点的相互作用进行验证;
a:四色杂交图像显示位点0(品红色),位点0的上游(位点2,黄色)或下游(位点3,绿色)59kb的斑点的在空间上局限于传统的bacfish(alexafluor594,红色);b,测得的b中tn5-fish的空间分辨率约为250nm;c,tn5-fish斑点之间空间距离的统计分析表明预测的e-p距离短于阴性对照。
具体实施方式
本发明涉及一种针对目标核酸靶标的探针的制备方法,该方法包括:
a)获取感兴趣的靶dna序列;
b)使用转座酶,将所述靶dna序列进行片段化的同时,在片段化的dna序列两端加上接头序列;和
c)利用所述接头序列,获取所述片段化的dna序列,以产生探针。
一个重要的优点在于,本方法不依赖或极少依赖于初始dna序列的特异性,能有效去除不想要的序列的区域(尤其是重复序列),从而使得本方法也不依赖于物种特异的cot-1dna对重复片段进行封闭;
一个重要的优点在于,本方法在制备探针时,所需的dna模板量约为50ng(例如30ng、35ng、40ng、45ng、55ng、60ng),远低于传统fish的1μg;同时,对于一个位点,仅需要做一次tn5高效转座酶的片段化之后,就可以进行大量的探针制备,流程简洁高效,而且性价比高;
一个重要的优点在于,本方法的实施要求操作者只需有基本的分子生物学技术,技术门槛较低;
一个重要的优点在于,本方法适合分析100kb及以内距离的染色质相互作用;
一个重要的优点在于,本方法具有至多大约1kb的基因组分辨率的标记能力。
本发明中靶dna序列可来源于任何含有靶dna的样品。
术语“样品”以其最广泛的含义使用。在一种含义中,其意指包括细胞(例如,人、细菌、酵母和真菌)、组织或活体、或获自任何来源的样本或培养物、以及生物样品。生物样品可以获自动物(包括人)、并且指其中发现的生物材料或组合物,包括但不限于骨髓、血液、血清、血小板、血浆、间隙液、尿液、脑脊液、核酸、dna、组织、及其纯化或过滤形式。然而,这些实例不应解释为限定可用于本发明的样品类型。
在一些实施方式中,所述样品为全基因组dna。
在一些实施方式中,所述转座酶是高活性的。
如本文所用,术语“核酸”指任何包含核酸的分子,包括但不限于dna或rna。该术语涵盖包含dna和rna的任何已知碱基类似物的序列,所述类似物包括但不限于:4-乙酰胞嘧啶、8-羟基-n6-甲基腺苷、吖丙啶基胞嘧啶、假异胞嘧啶、5-(羧基羟基甲基)尿嘧啶、5-氟尿嘧啶、5-溴尿嘧啶、5-羧甲基氨基甲基-2-硫代尿嘧啶、5-羧甲基氨基甲基尿嘧啶、二氢尿嘧啶、肌苷、n6-异戊烯基腺嘌呤、1-甲基腺嘌呤、1-甲基假尿嘧啶、1-甲基鸟嘌呤、1-甲基肌苷、2,2-二甲基鸟嘌呤、2-甲基腺嘌呤、2-甲基鸟嘌呤、3-甲基胞嘧啶、5-甲基胞嘧啶、n6-甲基腺嘌呤、7-甲基鸟嘌呤、5-甲基氨基甲基尿嘧啶、5-甲氧基氨基甲基-2-硫代尿嘧啶、β-d-甘露糖基q核苷、5’-甲氧基羰基甲基尿嘧啶、5-甲氧基尿嘧啶、2-甲硫基-n6-异戊烯基腺噪呤、尿啼啶-5-氧基乙酸甲酯、尿啼啶-5-氧基乙酸、氧基丁氧基胸苷(oxybutoxosine)、假尿嘧啶、q核苷、2-硫胞嘧啶、5-甲基-2-硫尿嘧啶、2-硫尿嘧啶、4-硫尿嘧啶、5-甲基尿嘧啶、n-尿嘧啶-5-氧基乙酸甲酯、尿嘧啶-5-氧基乙酸、假尿嘧啶、q核苷、2-硫胞嘧啶和2,6-二氨基嘌呤。
由本发明制备得到的探针一般用于检测的目标核酸为dna序列,但也不排除各种rna序列,或dna-rna混合序列,例如:由感兴趣的靶dna序列转录所得的mrna序列。
在一些实施方式中,所述感兴趣的靶dna序列为从初始序列中排除掉不想要的序列的区域得到。
如本文所用,术语“不想要的序列的区域”指实质上不含(例如85%、90%、91%、92%、93%、94%、95%、96%、97%、99%或100%不含)不想要的核酸的区域。不想要的核酸包括但不限于重复核酸、非保守序列、保守序列、富含gc的序列、富含at的序列、二级结构、非编码序列(例如启动子、增强子等)或编码序列。
在一些实施方式中,所述不想要的区域选自重复序列。
在一些实施方式中,所述排除的方法为扩增所述感兴趣的靶dna序列;
在一些实施方式中,所述扩增为pcr扩增。
在一些实施方式中,所述不想要的序列的区域至少为100bp;或为120bp、130bp、140bp、150bp、160bp、170bp、180bp、190bp、200bp、250bp、300bp、350bp、400bp、450bp、500bp、600bp、700bp、800bp、900bp、1000bp、1500bp、2000bp、3000bp、4000bp、5000bp、6000bp、7000bp、8000bp、9000bp、10000bp、20000bp、30000bp、40000bp、50000bp。
在一些实施方式中,所述转座酶选自tn1、tn2、tn3、tn4、tn5、tn6、tn7、tn9、tn10、tn551、tn971、tn916、tn1545、tn1681、tgf2、tol2、himar1以及harbi1中的一种或任意多种的组合。
tgf2和tol2来自于hat家族,himar1来自于tcl/mariner家族,harbi1来自于pif/harbinger家族。
在一些实施方式中,所述探针是标记的。
如本文所用的术语“标记”指可用于提供可检测的(优选可定量的)效果且可以连接至核酸或蛋白的任何原子或分子。标记包括但不限于染料;放射性标记,诸如32p;结合部分诸如生物素;半抗原诸如地高辛;发光、发磷光或发荧光部分;和单独的荧光染料或与可以通过荧光共振能量转移(fret)抑制或移动发射光谱的部分组合的荧光染料。标记可以提供可通过荧光、放射性、比色、重量测定、x射线衍射或吸收、磁性、酶活性等检测的信号。标记可以是带电荷的部分(正电荷或负电荷)或可选地,可以是电荷中性的。标记可以包括核酸或蛋白序列或由其组合,只要包含标记的序列是可检测的。在一些实施方案中,核酸在没有标记的情况下直接检测(例如,直接读取序列)。
在一些实施方式中,所述标记是荧光团、比色标记、量子点、生物素以及其他可以用于探测的标签分子(如用于拉曼衍射成像的炔烃基团,用于click反应的环烯烃,用于聚合物标记的引发集团),也可以选自多肽/蛋白分子,lna/pna,非天然氨基酸及其类似物(比如拟肽),非天然核酸及其类似物(拟核苷酸)和纳米结构(包括无机纳米颗粒,nv-center,聚集/组装诱导发光分子,稀土离子配体分子,多金属氧簇等)。
在一些实施方式中,所述标记是荧光团。
在一些实施方式中,所述荧光团可选自荧光素类染料、罗丹明类染料以及菁染料。
在一些实施方式中,所述荧光素类染料包括标准荧光素及其衍生物,如异硫氰酸荧光素(fitc)、羟基荧光素(fam)、四氯荧光素(tet)等。
在一些实施方式中,所述罗丹明类染料包括r101、四乙基罗丹明(rb200)和羧基四甲基罗丹明(tamra)等。
在一些实施方式中,所述菁染料主要选自两类,一类是噻唑橙(thiazoleorange,to)、噁唑橙(oxazoleorange,yo)系列及其二聚体染料,另一类是多甲川系列菁染料。
在一些实施方式中,荧光团还可以选择下述染料:二苯乙烯、萘酰亚胺、香豆素类、吖啶类、芘类等。
荧光团通常标记在引物或探针序列的5'端,但通过改变修饰键(例如-oh或-nh键)也可以将其置于3'端。
在一些实施方式中,在步骤c)中,产生探针的方法包括扩增、克隆、合成或其组合。
术语“扩增(ampiifying或amplification)”在“核酸”此用语的上下文中共同出现时,指产生多个拷贝的多核苷酸、或多核苷酸的部分,通常从少量的多核苷酸开始(例如,少至单个多核苷酸分子),其中扩增产物或扩增子通常是可检测的。多核苷酸的扩增包括多种化学和酶促方法。在聚合酶链反应(pcr)、滚环扩增(rca)或连接酶链反应(lcr)过程中从一个或几个拷贝的靶dna或模板dna分子产生多个dna拷贝是扩增的形式。扩增不限于起始分子的严格复制。例如,使用逆转录rt-pcr从样品中有限量的rna产生多个cdna分子是扩增的形式。此外,在转录过程期间从单一dna分子产生多个rna分子也是扩增的形式。
在一些实施方式中,在步骤c)中,产生探针的方法为:利用能够结合所述接头序列的引物,扩增所述片段化的dna序列。
术语“引物”指寡核苷酸,无论其是在经纯化的限制性消化物中天然存在或是合成产生的,所述寡核苷酸当置于诱导与核酸链互补的引物延伸产物合成的条件下时(例如,存在核苷酸和诱导剂诸如dna聚合酶和在合适的温度和ph下),所述寡核苷酸能够作为合成的起始点而起作用。引物优选是单链的,以用于扩增的最大效率,但可选地也可以是双链的。如果是双链的,则在用于制备延伸产物前,首先将引物处理以分开其链。优选地,引物是寡脱氧核糖核苷酸。引物应足够长,以在诱导剂存在的情况下引发延伸产物的合成。引物的精确长度将取决于许多因素,包括温度、引物来源和方法的使用。例如,在一些实施方案中,引物范围为10-100或更多个核苷酸(例如10-300、15-250、15-200、15-150、15-100、15-90、20-80、20-70、20-60、20-50个核苷酸等)。
在一些实施方案中,引物包含不与目标核酸杂交的另外的序列。术语“引物”包括化学修饰的引物、荧光修饰的引物、功能引物(融合引物)、序列特异性引物、随机引物、具有特异性和随机序列的引物、及dna和rna引物。
在一些实施方式中,所述引物是标记的。
在一些实施方式中,所述标记为上述术语“标记”所限定;
在一些实施方式中,所述标记选自荧光团、比色标记、量子点或生物素;优选荧光团。
根据本发明的再一方面,本发明还涉及进行杂交测定的方法,其包括利用如上所述的方法产生探针,并将靶核酸与所述探针接触。
如本文所用,术语“杂交”用于指互补核酸的配对。杂交和杂交强度(即核酸之间结合的强度)受此类因素如核酸之间的互补性程度、所涉及的条件的严格性、形成的杂交物的tm以及核酸内的g:c比率等的影响。在其结构内含有互补核酸的配对的单一分子将是“自杂交的”。
在一些实施方式中,所述杂交测定为原位杂交;
优选的,所述原位杂交为将所述探针与固定的目的细胞进行3dfish标记。
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例
一、材料和试剂
仪器:pcr仪(biored),杂交仪(abbottthermobite),水浴锅
试剂:rpmi1640培养基(购于gibco),dmem培养基(购于gibco),链霉素/青霉素双抗(购于gibco),胰蛋白酶(购于gibco),fbs(购于gibco)。基因组dna提取试剂盒(购于lifetechnology),qubitdna高灵敏度试剂盒(购于lifetechnology),antifade封片剂(含dapi,购于lifetechnology),fixogum(购于marubu),tn5转座酶试剂盒(购于vazyme),hs-taq(购于takara),pcr产物纯化试剂盒(购于zymo),37%盐酸(购于国药),tris-hcl(购于sigma),triton-x100(购于sigma),乙醇(购于sigma),硫酸葡聚糖(购于sigma),肝糖原(购于lifetechnology),20×ssc(购于lifetechnology),鲑鱼精子dna(购于lifetechnology),去离子甲酰胺(购于solarbio),pbs(购于solarbio),3m醋酸钠(购于solarbio),4%多聚甲醛(购于solarbio),np-40(购于solarbio),无dnase的rnasea(购于solarbio)。所有引物均由睿博兴科合成提供。本发明中涉及的所有bac克隆均购自lifetechnology。
细胞株:k562细胞(购于atcc),gm12878细胞(购于atcc),mouseesc细胞(购于atcc)。
耗材:superfrost载玻片(购于thermofisher),thermofisher1.5#盖玻片(购于thermofisher)。
二、探针制备方法步骤
1、扩增引物:针对需要标记的基因组位点,设计对应的引物,发给合成公司合成。荧光标记引物则根据tn5试剂盒提供的序列进行合成,所有荧光分子都标记在3’端。
2、基因组dna提取:对于每一种细胞,取1×106细胞,按照基因组dna提取试剂盒的实验步骤进行提取。提取后的dna用qubit定量,存于-20℃.
3、探针模板dna获取:取50nggenomicdna,将稀释好的引物加入pcr管,反应体积50微升进行pcr。pcr的条件为,98度3min,(98℃30s,55℃30s,72℃3min)×30循环,72度5min,4度保持。pcr产物经回收试剂盒纯化回收,qubit定量,存于-20℃。
4、tn5片段化:取50ng步骤3的dna产物,加入tn5酶以及反应buffer,总体积50μl。55度水浴锅处理10min,随后用pcr产物回收试剂盒纯化dna。
5、pcr扩增及荧光标记:步骤4所有产物投入pcr扩增。pcr的条件为,75度5min,(98度30s,55度30s,72度30s)×30循环,72度5min,4度保持。纯化dna产物后,取50ng作为模板,用带荧光标记的引物进行pcr扩增标记。pcr的条件为,98度3min,(98度30s,55度30s,72度30s)×30循环,72度5min,4度forever。标记后的产物用qubit定量后,加入2μl肝糖原和10μl鲑鱼精子dna进行2h的-80度乙醇沉淀(0.1倍体积3m醋酸钠,2.5倍体积无水乙醇)。醇沉后的探针用75%乙醇洗3次,挥发干净乙醇,并用杂交液(2×ssc,10%硫酸葡聚糖,50%去离子甲酰胺)重悬,存于-20度。
6、原位杂交:用4%多聚甲醛室温固定细胞10min,用0.1mtris-hcl洗10min,随后用含有10μg/mlrnasea的0.5%triton-x100透膜并消解rna,水浴37度处理30min,pbs洗3遍后用0.1m盐酸溶液室温处理30min;pbs洗3遍后,在50%去离子甲酰胺2×ssc溶液中室温处理30min,梯度乙醇脱水晾干。将10微升探针溶液(2ng/μl)与细胞混合后用fixogum封于玻片内,置杂交仪杂交(75度5min,37度过夜)。第二天用0.3%np-40的2×ssc溶液室温洗细胞3次,每次5min,随后用antifade封片剂封片,玻片边缘用fixogum封住溶液,避光保存4度或直接拍摄。
7、荧光成像及处理:将封好的片子用荧光显微镜或共聚焦显微镜进行拍摄。本发明中用的是蔡司的共聚焦显微镜(型号:lsm780),配备405,488,568,594和647激光及对应的滤光片组合,镜头为63×apoplanna1.4油镜。镜油为蔡司immersionoilf518,25度折射率1.515.图片采集软件为zensp2.3,处理软件为fiji(imagejcoreversion:1.52h)。
在一个具体的实施方式中,本发明公开了一种高分辨率荧光原位杂交探针的制备方法,包括下列步骤(如图1所示):
(1)基因组pcr获取探针模板:针对特定的标记片段,设计primer获取特定的dna片段,作为探针制备模板;
(2)探针制备模板dna的片段化:取特定量的探针制备模板dna(如1ng,5n,50ng,100ng,200ng,或500ng),加入tn5高活性转座酶进行片段化;
(3)pcr扩增:将步骤(2)获得的片段化后的dna进行pcr扩增,得到大量的无标记探针dna;
(4)探针的荧光标记:将步骤(3)获得的大量片段,用带荧光标记的引物进行pcr扩增,以加入荧光分子;
(5)原位杂交:将步骤(4)获得的荧光探针dna,与固定的目的细胞进行3dfish标记;
(6)荧光成像:将用该fish方法标记好的细胞,置于荧光显微镜下进行拍摄成像。
实施例1
将tn5-fish与传统bacfish联用,在wtmesc细胞和platr22-komesc细胞中,验证tn5-fish对基因组位点的标记特异性
实验结果如图2所示,在wtmesc中,两个距离6.9kb的临近基因组位点gm19705和platr22的tn5fish信号,均与覆盖两者区域的bacfish信号有良好的共定位;而在platr22-komesc细胞中,只有gm19705位点与bacfish信号有良好的共定位;而platr22位点的tn5-fish信号未检出,仅存在bacfish的信号。此实验说明了tn5fish标记具有很好的特异性。
实施例2
k562细胞中,tn5-fish和bacfish联用,验证100kb长度的染色质相互作用及1kb的基因组分辨率。
实验结果如图3所示,针对3个具有相互作用的基因位点,分别采用tn5-fish和bacfish验证其相互作用效果,可发现tn5-fish的信号与bacfish的信号有良好的共定位。与此同时,tn5-fish的探针模板dna长度为1kb,证明了tn5-fish具有在基因组中进行1kb分辨率的标记的能力,优于此前报道的多种fish方法的基因组分辨率(oligopaint分辨率为4kb,mb-fish为2.5kb,hd-fish为3.5kb,而casfish为10kb)。
实施例3
在gm12878细胞中,利用多色tn5-fish,对预测的位于chr2:227672028-227743852处两端位点的相互作用,进行验证
根据chip-seq数据和hic数据,我们对gm12878细胞中的位于chr2:227672028-227743852处两端位点的两个相距59kb的存在e-p相互作用的位点(site1和site2)进行了验证,并用反向相同距离的位点(site3)作为对照,同时配备了能够同时覆盖3个位点的bac探针作为参考。如图4所示,3个位点分别用2kb长度的片段制备了3种颜色的tn5fish探针,三者进行杂交后,在细胞内的信号点均相互聚集在一起。选取每个位点的荧光标记信号中心点,进行两者之间空间距离的测量后发现,site1至site2的距离分布小于site1至site3的空间距离,两者具有显著性差异,说明tn5十分适合分析100kb及以内距离的染色质相互作用,而这个距离往往不适合用传统的bacfish进行标记和分析。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!