一种具有杀菌活性的咪唑类药物分子及其制备方法与流程
本发明属于抗菌药物合成技术领域,具体涉及一种具有杀菌活性的咪唑类药物分子及其制备方法。
背景技术:
含氮杂环化合物一直是含能材料研究中的一个热点。咪唑环是一种重要的五元含氮杂环化合物,在许多天然产物和具有药理学活性的化合物中都可看到咪唑片段。许多取代咪唑衍生物还可以作为植物生长调节剂,p38MAP和B.Raf激酶抑制剂,胰高血糖素受体。此外,Losartan,Candesartan,Eprosartant等药物中都含有多官能化的咪唑环。鉴于咪唑类化合物的广泛应用,高效地合成多取代咪唑对拓展咪唑类衍生物的应用具有重要的意义。
早在1858年,德国科学家Heinrich Debus利用乙二酮、甲醛、胺首次合成了三取代咪唑环衍生物,但其产率较低;2008年,Kaila从脒硫脲出发,在二氯化汞的作用下先与伯胺反应生成脒基胍,再在DBU碱性条件下与溴代酮化合物发生反应12h,生成1,2,4,5-四取代咪唑化合物,该反应时间较长,而且使用的二氯化汞催化剂毒性较大;2017年,Yu等设计了一种N取代2-氨基乙腈原料,在钯催化剂下与硼酸作用构建C-C偶联和C-N缩合串联反应,合成了1,2,4,5-四取代咪唑化合物,钯催化剂价格昂贵,工业化生产成本较高。鉴于三唑并苯并二氮杂类衍生物以及咪唑衍生物在医药上的广泛应用,合成此两类杂环化合物具有重要的意义。通过文献调研发现,有大量合成此两类化合物的方法被报道,但有的步骤繁琐,有的要用到昂贵催化剂,有的副产物多,因此,探索具有步骤简单、条件温和、原子经济性高的合成方法变得十分重要。
由于工作性质的原因。医院污水在无害化处理前含有大量的致病微生物、化学有害物及放射性污染物等,对周围环境具有极大的危害性,是引起水源污染与传染病暴发流行的潜在传染源。对医疗机构排放的污水开展监测工作是医疗卫生的重要工作之一。研究发现,医院无水中含有大量的大肠杆菌和金黄色葡萄球菌,因此研究对这两个细菌抑制性好的杀菌药物对处理医院无水很具有重要意义。
本发明以对甲氧基苯甲酸为起始原料,经过酰胺化、羟醛缩合、分子内环合等五步反应得到一种结构新颖的咪唑类化合物,并对大肠杆菌和金黄葡萄球菌进行了抗菌活性测试。
技术实现要素:
本发明解决的技术问题是提供了一种具有杀菌活性的咪唑类药物分子及其制备方法。
本发明为解决上述技术问题采用如下技术方案,一种具有杀菌活性的咪唑类药物分子,其特征在于该化合物具有如下结构:
本发明为解决上述技术问题采用如下技术方案,一种具有杀菌活性的咪唑类药物分子的制备方法,其特征在于具体步骤为:
(1)、对甲氧基苯甲酸与氨基乙酸乙酯在缩合试剂作用下反应得到N-乙酸乙酯基-对甲氧基苯甲酰胺;
(2)、N-乙酸乙酯基-对甲氧基苯甲酰胺与劳森试剂反应得到N-乙酸乙酯基-对甲氧基硫代苯甲酰胺;
(3)、N-乙酸乙酯基-对甲氧基硫代苯甲酰胺与碘甲烷反应得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯;
(4)、2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯与甲酰胺缩合后环合,再水解得到2-(4-甲氧基苯基)-1H-咪唑-4-羧酸;
(5)、2-(4-甲氧基苯基)-1H-咪唑-4-羧酸与间甲基苯乙醇发生酯化反应得到目标化合物。
进一步限定,步骤(1)的具体过程为:把对甲氧基苯甲酸、氨基乙酸乙酯和一定量的缩合试剂加入到一定溶剂中,搅拌均匀后在氮气保护下,室温条件下反应至对甲氧基苯甲酸反应完全,向反应液中加入饱和氯化钠溶液洗涤反应液,搅拌均匀后分出有机相,水相加入冰乙酸调节pH至中性,再用二氯甲烷萃取多次,合并有机相,减压浓缩,将剩余物经过硅胶柱层析进行纯化得到N-乙酸乙酯基-对甲氧基苯甲酰胺;所述的缩合试剂为O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯或1-羟基苯并三唑;所述的溶剂为二氯甲烷或乙腈;所述对甲氧基苯甲酸与氨基乙酸乙酯与缩合试剂的投料量摩尔比为1:1.1:1~1.5。
进一步限定,步骤(2)的具体过程为:把一定量的N-乙酸乙酯基-对甲氧基苯甲酰胺和劳森试剂加入到反应溶剂中,加热到一定温度,在通风系统良好的条件下减压浓缩,然后加入饱和氯化钠溶液,搅拌均匀后再加入二氯甲烷,继续搅拌一段时间,分出有机相,将剩余物经过柱层析进行纯化得到N-乙酸乙酯基-对甲氧基硫代苯甲酰胺;所述的反应溶剂为四氢呋喃或甲苯;所述的N-乙酸乙酯基-对甲氧基苯甲酰胺与劳森试剂的投料量摩尔比为1:0.7;所述的反应温度为60~80℃。
进一步限定,步骤(3)的具体过程为:把N-乙酸乙酯基-对甲氧基硫代苯甲酰胺和一定量的碱性化合物加入无水乙醇,在一定反应温度缓慢滴加溶有碘甲烷的乙醇溶液,滴加完后搅拌反应一段时间,用蒸馏水洗涤混合液,再分别用乙酸乙酯萃取多次,合并有机相,有机相经过饱和氯化钠溶液洗涤两次,分离有机相,将得到的有机相用无水硫酸钠干燥,浓缩,剩余物经过硅胶柱层析进行纯化得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯;所述的碱性化合物为甲醇钠、乙醇钠或叔丁醇钾;所述的N-乙酸乙酯基-对甲氧基硫代苯甲酰胺与碱性化合物与碘甲烷的投料量摩尔比为1:1~1.2:3;所述的反应温度为-15~-10℃。
进一步限定,步骤(4)的具体过程为:把带有搅拌的反应瓶中至于-60℃条件下,氮气保护条件下,把一定量的2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯和甲酰胺加入到四氢呋喃中,再滴加溶有一定量催化剂的四氢呋喃溶液,于-60℃条件反应至原料反应完全后,恢复至室温,分别用乙酸乙酯萃取多次,合并有机相,有机相减压浓缩;在0℃条件下,向浓缩物中,滴加2N的盐酸调节反应液pH为2左右,加入氯化钠和水,再加入碳酸氢钠调节反应液pH为6到7,滴加质量分数为10%的氢氧化钠溶液,室温反应,pH不超过8;TLC监控原料反应完全,降温至-10℃,搅拌反应均匀后抽滤,得到2-(4-甲氧基苯基)-1H-咪唑-4-羧酸;所述的催化剂为二(三甲基硅基)氨基钠,双-(三甲基硅基)胺锂或双-(三甲基硅基)氨基钾;所述的2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯与甲酰胺与催化剂的投料量摩尔比为1:2:2。
进一步限定,步骤(5)的具体过程为:在反应瓶中加入2-(4-甲氧基苯基)-1H-咪唑-4-羧酸和二氯亚砜,在室温条件下搅拌反应一段时间,然后再真空条件下蒸除未反应的二氯亚砜,再加入二氯甲烷,在10℃条件下,向反应液中滴加溶有一定量的间甲基苯乙醇的二氯甲烷溶液,滴加完后继续反应至原料反应完全,向反应液中加入水,搅拌均匀后,分出有机相,水相再用二氯甲烷萃取多次,合并有机相,浓缩后得在甲醇中重结晶得到目标化合物;所述的2-(4-甲氧基苯基)-1H-咪唑-4-羧酸与间甲基苯乙醇的投料量摩尔比为1:1。
本发明具有的有益效果:本发明通过新方法合成了一种结构新颖的咪唑类药物分子,并通过微量二倍稀释法进行抗菌活性测试发现目标化合物具有良好的抗菌作用。
附图说明
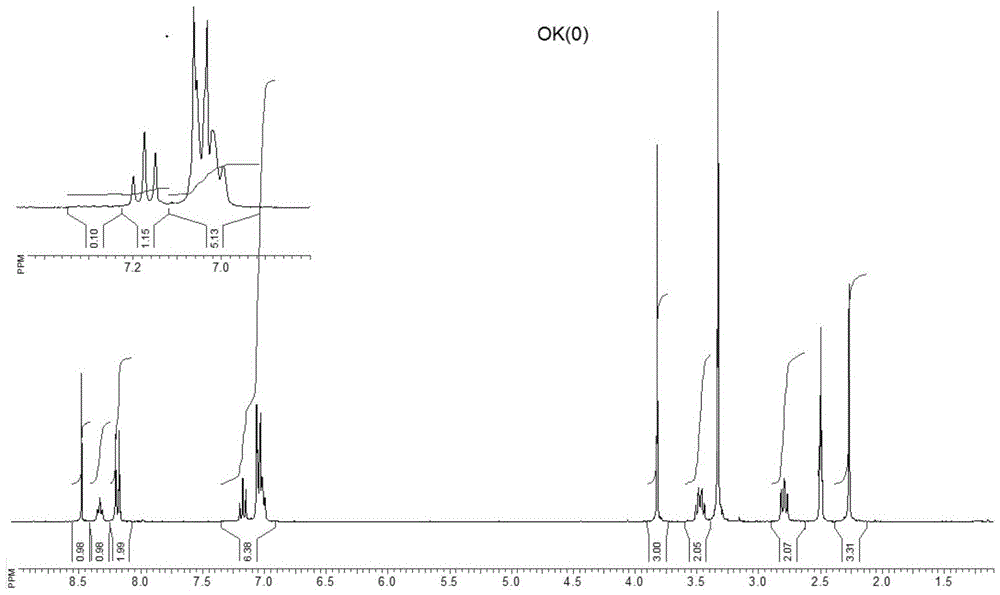
图1目标化合物的核磁氢谱
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
在带有搅拌器的反应瓶中,把对甲氧基苯甲酸15g、氨基乙酸乙酯11g和O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯48g加入到二氯甲烷300mL中,搅拌均匀后在氮气保护下,室温条件下反应3.5h,向反应液中加入饱和氯化钠溶液200mL洗涤反应液,搅拌10min后分出有机相,水相加入冰乙酸调节pH至中性,再用二氯甲烷100mL萃取多次,合并有机相,减压浓缩,将剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=5:1)得到N-乙酸乙酯基-对甲氧基苯甲酰胺12.1g;1H NMR(400MHz,DMSO-d6):δ8.47(s,1H),7.74(d,J=8.0Hz,2H),6.93(dd,J1=8.0Hz,J2=4.0Hz,2H),4.20-4.14(m,4H),3.77(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H).13C NMR(101MHz,DMSO-d6):δ171.1,166.9,162.4,128.2,125.7,113.8,61.5,54.8,41.7,14.1。
实施例2
在带有搅拌器的反应瓶中,把对甲氧基苯甲酸15g、氨基乙酸乙酯11g和O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯48g加入到乙腈300mL中,搅拌均匀后在氮气保护下,室温条件下反应2h,向反应液中加入饱和氯化钠溶液200mL洗涤反应液,搅拌10min后分出有机相,水相加入冰乙酸调节pH至中性,再用二氯甲烷200mL萃取多次,合并有机相,减压浓缩,将剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=5:1)得到N-乙酸乙酯基-对甲氧基苯甲酰胺16.5g;1H NMR(400MHz,DMSO-d6):δ8.47(s,1H),7.74(d,J=8.0Hz,2H),6.93(dd,J1=8.0Hz,J2=4.0Hz,2H),4.20-4.14(m,4H),3.77(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H).13C NMR(101MHz,DMSO-d6):δ171.1,166.9,162.4,128.2,125.7,113.8,61.5,54.8,41.7,14.1。
实施例3
在带有搅拌器的反应瓶中,把对甲氧基苯甲酸15g、氨基乙酸乙酯11g和EDC·HCl 20g和1-羟基苯并三唑20g加入到乙腈400mL中,搅拌均匀后在氮气保护下,室温条件下反应5.2h,TLC监控对甲氧基苯甲酸反应完全,向反应液中加入饱和氯化钠溶液200mL洗涤反应液,搅拌10min后分出有机相,水相加入冰乙酸调节pH至中性,再用二氯甲烷200mL萃取多次,合并有机相,减压浓缩,将剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=5:1)得到N-乙酸乙酯基-对甲氧基苯甲酰胺21.2g;1H NMR(400MHz,DMSO-d6):δ8.47(s,1H),7.74(d,J=8.0Hz,2H),6.93(dd,J1=8.0Hz,J2=4.0Hz,2H),4.20-4.14(m,4H),3.77(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H).13C NMR(101MHz,DMSO-d6):δ171.1,166.9,162.4,128.2,125.7,113.8,61.5,54.8,41.7,14.1。
实施例4
在带有搅拌器的反应瓶中,把对甲氧基苯甲酸15g、氨基乙酸乙酯11g和EDC·HCl 20g和1-羟基苯并三唑13g加入到乙腈400mL中,搅拌均匀后在氮气保护下,室温条件下反应5.2h,向反应液中加入饱和氯化钠溶液200mL洗涤反应液,搅拌10min后分出有机相,水相加入冰乙酸调节pH至中性,再用二氯甲烷200mL萃取多次,合并有机相,减压浓缩,将剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=5:1)得到N-乙酸乙酯基-对甲氧基苯甲酰胺17.7g;1H NMR(400MHz,DMSO-d6):δ8.47(s,1H),7.74(d,J=8.0Hz,2H),6.93(dd,J1=8.0Hz,J2=4.0Hz,2H),4.20-4.14(m,4H),3.77(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H).13C NMR(101MHz,DMSO-d6):δ171.1,166.9,162.4,128.2,125.7,113.8,61.5,54.8,41.7,14.1。
实施例5
在反应瓶中,把N-乙酸乙酯基-对甲氧基苯甲酰胺24g和劳森试剂28g加入到四氢呋喃200mL,加热回流反应2.5h,在通风系统良好的条件下减压浓缩(劳森试剂副产物具有恶臭),然后加入饱和氯化钠溶液130mL,搅拌均匀后再加入二氯甲烷100mL,继续搅拌10min,分出有机相,将剩余物经过柱层析进行纯化(硅胶柱,洗脱剂,石油醚:乙酸乙酯=3:1),得到N-乙酸乙酯基-对甲氧基硫代苯甲酰胺18.5g;MS(ESI)m/z:254.3(M+H+)。
实施例6
在反应瓶中,把N-乙酸乙酯基-对甲氧基苯甲酰胺24g和劳森试剂28g加入到甲苯200mL,加热至80℃反应1h,在通风系统良好的条件下减压浓缩(劳森试剂副产物具有恶臭),然后加入饱和氯化钠溶液130mL,搅拌均匀后再加入二氯甲烷100mL,继续搅拌10min,分出有机相,将剩余物经过柱层析进行纯化(硅胶柱,洗脱剂,石油醚:乙酸乙酯=3:1),得到N-乙酸乙酯基-对甲氧基硫代苯甲酰胺21.6g;MS(ESI)m/z:254.3(M+H+)。
实施例7
在带有搅拌器的反应瓶中,把N-乙酸乙酯基-对甲氧基硫代苯甲酰胺25g和乙醇钠7g加入无水乙醇180mL,反应体系温度降至-15~-10℃,缓慢滴加溶有碘甲烷43g的乙醇溶液200mL,滴加完后搅拌反应3h,用蒸馏水100mL洗涤混合液,再分别用乙酸乙酯60mL萃取三次,合并有机相,有机相经过饱和氯化钠50mL溶液洗涤两次,分离有机相,将得到的有机相用无水硫酸钠干燥,浓缩,剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=7:1),得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯19.4g;1H NMR(400MHz,DMSO-d6):δ7.71(d,J=8.0Hz,2H),6.97(dd,J1=8.0Hz,J2=4.0Hz,2H),4.26(s,2H),4.19(dd,J1=8.0Hz,J2=8.0Hz,2H),3.72(s,3H),2.61(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H),MS(ESI)m/z:268.4(M+H+)。
实施例8
在带有搅拌器的反应瓶中,把N-乙酸乙酯基-对甲氧基硫代苯甲酰胺25g和甲醇钠5.5g加入无水乙醇180mL,反应体系温度降至-15~-10℃,缓慢滴加溶有碘甲烷43g的乙醇溶液200mL,滴加完后搅拌反应3h,用蒸馏水100mL洗涤混合液,再分别用乙酸乙酯60mL萃取三次,合并有机相,有机相经过饱和氯化钠50mL溶液洗涤两次,分离有机相,将得到的有机相用无水硫酸钠干燥,浓缩,剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=7:1),得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯14.9g;1H NMR(400MHz,DMSO-d6):δ7.71(d,J=8.0Hz,2H),6.97(dd,J1=8.0Hz,J2=4.0Hz,2H),4.26(s,2H),4.19(dd,J1=8.0Hz,J2=8.0Hz,2H),3.72(s,3H),2.61(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H),MS(ESI)m/z:268.4(M+H+)。
实施例9
在带有搅拌器的反应瓶中,把N-乙酸乙酯基-对甲氧基硫代苯甲酰胺25g和叔丁醇钾11g加入无水乙醇180mL,反应体系温度降至-15~-10℃,缓慢滴加溶有碘甲烷43g的乙醇溶液200mL,滴加完后搅拌反应3h,TLC监控原料反应完全,用蒸馏水100mL洗涤混合液,再分别用乙酸乙酯60mL萃取三次,合并有机相,有机相经过饱和氯化钠50mL溶液洗涤两次,分离有机相,将得到的有机相用无水硫酸钠干燥,浓缩,剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=7:1),得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯22.5g;1H NMR(400MHz,DMSO-d6):δ7.71(d,J=8.0Hz,2H),6.97(dd,J1=8.0Hz,J2=4.0Hz,2H),4.26(s,2H),4.19(dd,J1=8.0Hz,J2=8.0Hz,2H),3.72(s,3H),2.61(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H),MS(ESI)m/z:268.4(M+H+)。
实施例10
在带有搅拌器的反应瓶中,把N-乙酸乙酯基-对甲氧基硫代苯甲酰胺25g和叔丁醇钾13.5g加入无水乙醇180mL,反应体系温度降至-15~-10℃,缓慢滴加溶有碘甲烷43g的乙醇溶液200mL,滴加完后搅拌反应3h,TLC监控原料反应完全,用蒸馏水100mL洗涤混合液,再分别用乙酸乙酯60mL萃取三次,合并有机相,有机相经过饱和氯化钠50mL溶液洗涤两次,分离有机相,将得到的有机相用无水硫酸钠干燥,浓缩,剩余物经过硅胶柱层析进行纯化(洗脱剂,石油醚:乙酸乙酯=7:1),得到2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯25.1g;1H NMR(400MHz,DMSO-d6):δ7.71(d,J=8.0Hz,2H),6.97(dd,J1=8.0Hz,J2=4.0Hz,2H),4.26(s,2H),4.19(dd,J1=8.0Hz,J2=8.0Hz,2H),3.72(s,3H),2.61(s,3H),1.24(t,J1=4.0Hz,J2=4.0Hz,3H),MS(ESI)m/z:268.4(M+H+)。
实施例11
把带有搅拌的反应瓶中至于-60℃条件下,氮气保护条件下,把2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯27g和甲酰胺9g加入到四氢呋喃120mL中,再滴加溶有二(三甲基硅基)氨基钠37g的四氢呋喃溶液200mL,于-60℃条件反应7h,TLC监控原料反应完全后,恢复至室温,分别用乙酸乙酯120mL萃取三次,合并有机相,有机相减压浓缩;在0℃条件下,向浓缩物中,滴加2N的盐酸调节反应液pH为2左右,加入氯化钠20g,再加入水250mL,加入碳酸氢钠调节反应液pH为6到7,滴加质量分数为10%的氢氧化钠溶液,室温反应,pH不超过8;TLC监控原料反应完全,降温至-10℃,搅拌反应1h后抽滤,得到2-(4-甲氧基苯基)-1H-咪唑-4-羧酸13.6g;元素分析计算值[C11H10N2O3]:C,60.55;H,4.62;N,12.84.实测值:C,60.39;H,4.71;N,12.75。
实施例12
把带有搅拌的反应瓶中至于-60℃条件下,氮气保护条件下,把2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯27g和甲酰胺9g加入到四氢呋喃120mL中,再滴加溶有双-(三甲基硅基)胺锂33g(0.2mol)的四氢呋喃溶液200mL,于-60℃条件反应7h,TLC监控原料反应完全后,恢复至室温,分别用乙酸乙酯120mL萃取三次,合并有机相,有机相减压浓缩;在0℃条件下,向浓缩物中,滴加2N的盐酸调节反应液pH为2左右,加入氯化钠20g,再加入水250mL,加入碳酸氢钠调节反应液pH为6到7,滴加质量分数为10%的氢氧化钠溶液,室温反应,pH不超过8;TLC监控原料反应完全,降温至-10℃,搅拌反应1h后抽滤,得到2-(4-甲氧基苯基)-1H-咪唑-4-羧酸19.7g;元素分析计算值[C11H10N2O3]:C,60.55;H,4.62;N,12.84.实测值:C,60.39;H,4.71;N,12.75。
实施例13
把带有搅拌的反应瓶中至于-60℃条件下,氮气保护条件下,把2-(((4-甲氧基苯)(甲硫基)(亚甲基)(氨基)醋酸乙酯27g和甲酰胺9g加入到四氢呋喃120mL中,再滴加溶有双-(三甲基硅基)氨基钾40g(0.2mol)的四氢呋喃溶液200mL,于-60℃条件反应7h,TLC监控原料反应完全后,恢复至室温,分别用乙酸乙酯120mL萃取三次,合并有机相,有机相减压浓缩;在0℃条件下,向浓缩物中,滴加2N的盐酸调节反应液pH为2左右,加入氯化钠20g,再加入水250mL,加入碳酸氢钠调节反应液pH为6到7,滴加质量分数为10%的氢氧化钠溶液,室温反应,pH不超过8;TLC监控原料反应完全,降温至-10℃,搅拌反应1h后抽滤,得到2-(4-甲氧基苯基)-1H-咪唑-4-羧酸15.3g;元素分析计算值[C11H10N2O3]:C,60.55;H,4.62;N,12.84.实测值:C,60.39;H,4.71;N,12.75。
实施例14
在反应瓶中加入2-(4-甲氧基苯基)-1H-咪唑-4-羧酸22g和二氯亚砜100mL,在室温条件下搅拌反应1h,然后再真空条件下蒸除未反应的二氯亚砜,再加入二氯甲烷130mL,在10℃条件下,向反应液中滴加溶有间甲基苯乙醇14g的二氯甲烷溶液100mL,滴加完后继续反应30min,TLC监控原料反应完全,向反应液中加入水100mL,搅拌均匀后,分出有机相,水相再用二氯甲烷50mL萃取多次,合并有机相,浓缩后得在甲醇中重结晶得到目标化合物27.5g;1H NMR(400MHz,DMSO-d6):δ8.49(s,1H,NH-1H),8.37(s,1H,CH-H),8.17(d,J=8.0Hz,2H,Ar-2H),7.18(t,J1=8.0Hz,J2=8.0Hz,1H,Ar-1H),7.07-6.99(m,5H,Ar-5H),3.81(s,3H,OCH3-3H),3.46(dd,J1=8.0Hz,J2=4.0Hz,2H,CH2-2H),2.79(dd,J1=8.0Hz,J2=4.0Hz,2H,CH2-2H),2.28(s,3H,CH3-3H).MS(ESI)m/z:337.5(M+H+);元素分析计算值[C20H20N2O3]:C,71.41;H,5.99;N,8.33.实测值:C,71.09;H,5.93;N,8.51。
实施例15
抗菌活性测试
以诺氟沙星为对阳性照,采用微量二倍稀释法测定目标化合物对金黄色葡萄球菌和大肠杆菌抗菌活性。将待测的化合物溶解在二甲基亚砜中,用微量移液器在96孔培养板上二倍梯度稀释样品,得到0.125~128ug/mL的不同质量浓度溶液,加入菌液,细菌浓度为5×105CFU/mL。在37℃下培养24h,记录结果,若有菌生长孔板底部会有白色沉淀,96孔板上横排中未有沉淀的最后一孔所对应的浓度便为该药的MIC值。
由上表可以看出,目标化合物对大肠杆菌的作用效果由于金黄葡萄球菌的作用效果。
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!