一种双碱基突变的高活性sgRNA骨架、sgRNA骨架载体及其应用的制作方法
本发明属于生物技术领域,涉及一种载体,具体涉及一种双碱基突变的高活性sgrna骨架、sgrna骨架载体及其应用。
背景技术:
crispr/cas9系统是第三代基因编辑技术,依赖sgrna引导cas9核酸酶在特定位置进行基因编辑,被广泛用于基因组编辑,在农业、生命科学以及医学领域发挥重要的价值。crispr/cas9系统主要包括两个部分:(1)来自链球菌(s.pyogenescas9(spcas9))的核酸酶;(2)singleguiderna(sgrna),sgrna表达载体包含20bp特异性可变靶位点和82bp固定不变的sgrna骨架(sgrnascaffold)。sgrna骨架是人为构建的crispr靶向识别系统中两个rna分子(crisprrna(crrna)、trans-activatingcrisprrna(tracrrna))的嵌合体(jinek,etal.,2012;hsupd,etal.,2013)。
crrna是全长为42bp的rna,其5’端20bp的碱基可以和基因组目标序列(targetsequence)匹配,其随着目标基因改变。crrna的3’端为22bp不可变序列,即“repeat”序列,用于和tracrrna配对。tracrrna是全长约为87bp的rna序列,其5’可以和crrna的3’的“repeat-sequence”配对,称为“anti-repeat”,而剩余序列可以形成多个有发卡环的二级结构。sgrna就是将原来单独的crrna和tracrrna人工嵌合成单一的导向rna,使其可以发挥原来crrna和tracrrna的作用又方便之后的实验操作。目前广泛应用的全长82bp的sgrna骨架序列来自张峰实验室(ranfa,etal.,2013;hsud,etal.,2013),该sgrna骨架也是经过多个团队多次优化的。2012年末,jennifera.doudna和emmanuellecharpentier两个团队合作首次提出将crrna和tracrrna连接起来成为一个rna,即sgrna(jinek,etal.,2012),这就是现在通用sgrna骨架的原型。该sgrna骨架将crrna的3’端10个碱基以及tracrrna的5’碱基19个碱基删除,并且将tracrrna的3’尾巴切掉一部分,通过gaaa4个碱基将剩下的crrna和tracrrna连接起来。该sgrna后来被张峰团队证明在体内介导cas9酶的基因编辑效果不够理想。之后2013年初,张峰团队和georgem.church分别在同一期science杂志对jennifera.doudna的sgrna进行优化,通过加上tracrrna的3’尾巴,使得该sgrna介导的cas9对目标基因的编辑效果更好更稳定(congl,etal.,2013、malip,etal.,2013)。2013年6月份,张峰团队又再次对该sgrna进行评估,认为tracrrna尾巴对于体内spcas9的最佳表达和活性至关重要(hsupd,etal.,2013)。至此,sgrnascaffold序列就已成型,其包括12bpcrrna,66bptracrrna以及人工添加的4个用于连接crrna和tracrrna的碱基gaaa。sgrna骨架任何序列都很关键,对于cas9酶的活性介导至关重要,对cas9核酸酶复合体切割dna是必需的。
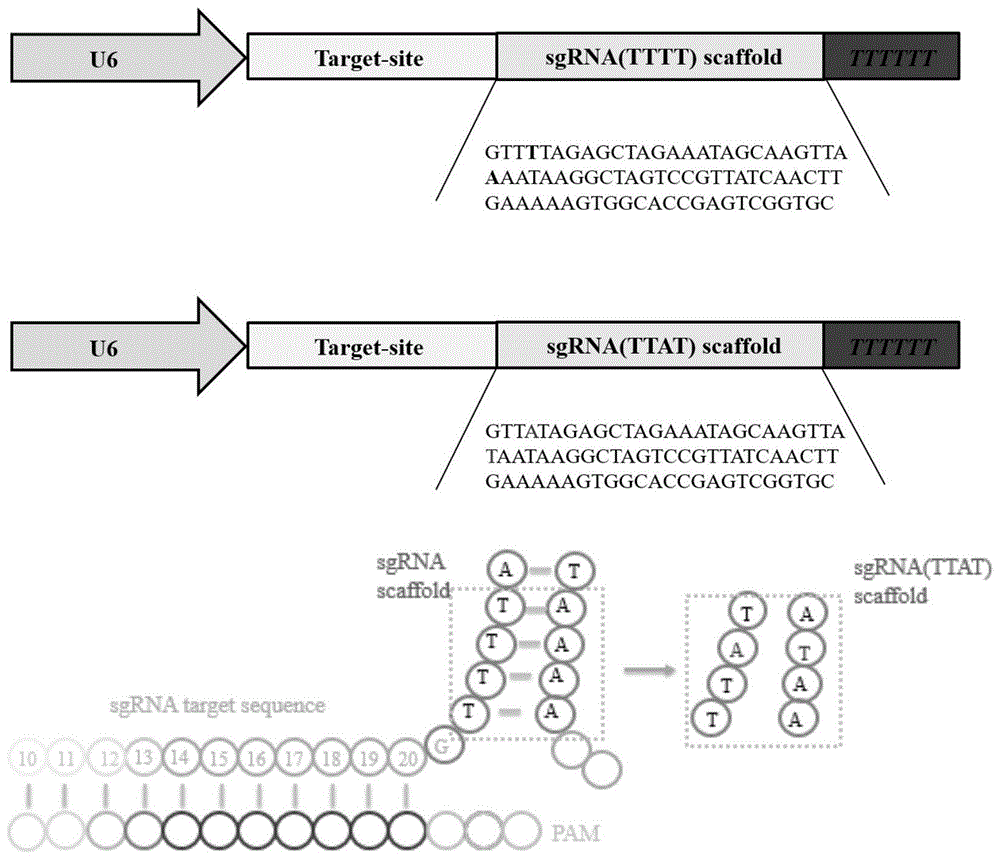
sgrna表达载体以u6启动子启动sgrna序列的表达。u6启动子是rna聚合酶iii依赖的启动子,在哺乳动物细胞内引导rna的转录,u6启动子具有明确的转录起始点g,以连续的5个t序列终止转录,转录产物会在3′端悬挂2-4个寡聚u(linx,etal.2004),且遇到连续的4-5个tttt也会终止转录。在张峰实验组sgrnascaffold序列中,除过序列末端有5个连续的t碱基,在sgrnascaffold起始位点即crrna的repeat序列也有4个连续的t碱基,申请人推测该序列会影响转录提前终止,从而降低sgrnascaffold转录,也就间接影响cas9/sgrna复合体的基因编辑活力。张峰团队为了解决该问题而将tttt改为tatt碱基,但是通过t7endonucleaseⅰ酶切鉴定,结果显示,该突变对其介导cas9的编辑活力几乎没有提高(hsupd,etal.,2013)。
技术实现要素:
针对现有技术中存在的问题,本发明提供一种双碱基突变的高活性sgrna骨架、sgrna骨架载体及其应用,显著提高crispr/cas9系统的基因编辑效率。
本发明是通过以下技术方案来实现:
一种双碱基突变的高活性sgrna骨架,将其命名为sgrna(ttat)scaffold,其核苷酸序列如seq.id.no.1所示。
一种双碱基突变的高活性sgrna骨架载体,将其命名为pu6-sgrna(ttat)scaffold,其核苷酸序列如seq.id.no.4所示。
一种靶向目标基因的高活性sgrna骨架载体,是在上述pu6-sgrna(ttat)scaffold上,利用粘性末端连入靶向目标基因的sgrna片段,获得的载体命名为pu6-sgrna(ttat)-targetsite。
优选的,目标基因为人tpi1p2基因、人emx1基因或者人rankl基因。
进一步的,靶向人tpi1p2基因的sgrna片段的核苷酸序列如seq.id.no.5所示。
进一步的,靶向人emx1基因的sgrna片段的核苷酸序列如seq.id.no.6所示。
进一步的,靶向人rankl基因的sgrna片段的核苷酸序列如seq.id.no.7所示。
所述的靶向目标基因的高活性sgrna骨架载体在基因组编辑中的应用。
与现有技术相比,本发明具有以下有益的技术效果:
本发明通过改变现有sgrna骨架序列中的两个碱基,即原sgrna骨架序列crrnarepeat序列中连续的tttt碱基替换为ttat碱基,并将与其对应互补的trancrna序列中连续aaaa碱基替换为ataa碱基,形成新的sgrna骨架,命名为sgrna(ttat)scaffold。实验证明sgrna(ttat)scaffold可以提高sgrna转录水平及其介导的cas9酶切活性,能够使sgrna引导的crispr/cas9系统在进行基因编辑和转录调控时,活性增强。这种方法简便易操作,只通过两个碱基的改变就能显著增强sgrnascaffold的转录水平,从而提高crispr/cas9系统打靶效率,提高基因编辑效率。
本发明所构建的包含sgrna(ttat)scaffold的高活性sgrna骨架载体,能有效介导cas9蛋白的基因组编辑作用,且高于普通sgrna载体介导的编辑效果。
本发明构建的高活性sgrna骨架载体pu6-sgrna(ttat)-targetsite与载体hcas9共转染hek293细胞,进行了对目标基因的编辑实验,同时将已有通用sgrna骨架载体与载体hcas9共转染hek293细胞作对照。实验结果表明,本发明的高活性sgrna的载体与hcas9载体共转染细胞后,对目标基因可以进行有效编辑,且显著高于已有通用sgrna载体介导的编辑效果。
附图说明
图1是sgrnascaffold和sgrna(ttat)scaffold序列结构图。
图2是pu6-sgrnascaffold和pu6-sgrna(ttat)scaffold载体结构图。
图3是pu6-sgrna-targetsite和pu6-sgrna(ttat)-targetsite载体结构图。
图4是实施例1构建的pu6-sgrna(ttat)-tpi1p2载体介导的目标基因编辑效率检测图。
图5是实施例2构建的pu6-sgrna(ttat)-emx1载体介导的目标基因编辑效率检测图。
图6是实施例3构建的pu6-sgrna(ttat)-rankl载体介导的目标基因编辑效率检测图。
具体实施方式
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
本发明的高活性sgrna骨架载体是在张峰实验室的sgrnascaffold载体的基础上通过对该sgrna骨架序列两个碱基的改变实现的。pcr获得的高活性骨架片段sgrna(ttat)scaffold,通过ecorⅰ与speⅰ位点连入载体puc19/hu6-sgrnascaffold中,替换原载体的sgrnascaffold获得高活性sgrna骨架载体pu6-sgrna(ttat)scaffold(如图1和图2);使用bsaⅰ酶切载体pu6-sgrna(ttat)scaffold,并利用粘性末端连入针对目标基因的sgrna序列,获得载体pu6-sgrna(ttat)-targetsite(如图3)。所述粘性末端连入针对目标基因编辑靶点的长度为19或20个核苷酸,退火构成其sgrna片段的引物序列,所述的针对目标基因的sgrna序列是针对目标基因编辑靶点,它们是针对tpi1p2基因、emx1基因或者rankl基因启动子中的任意一种,但并不只限于这三种。
本发明构建方法,具体步骤为:
(1)pcr合成高活性sgrna骨架片段sgrna(ttat)scaffold;
根据已知sgrna骨架序列,以骨架上游ecorⅰ位点为5’起点向下游即3’方向延伸合成上游引物59bp,该上游引物将原sgrna骨架序列crrnarepeat序列中连续的tttt碱基替换为ttat碱基,并将与其对应互补的trancrna序列中连续aaaa碱基替换为ataa碱基,即改变原sgrna骨架两个碱基。以该已知骨架末端下游spei位点为5’起点并加上保护碱基向上游即3’方向延伸合成下游引物59bp,上下游引物之间有16bp碱基可以互相匹配。pcr扩增形成5’末端带有ecorⅰ位点,3’末端带有speⅰ位点的长度与原sgrnascaffold长度相同的高活性sgrna(ttat)scaffold片段。
(2)构建pu6-sgrna(ttat)scaffold载体
将高活性sgrna(ttat)scaffold片段经过ecorⅰ与speⅰ酶切,使用1%的琼脂糖凝胶电泳后回收片段,载体puc19/hu6-sgrnascaffold通过ecorⅰ与speⅰ酶切后,使用1%的琼脂糖凝胶电泳后回收载体;使用t4连接酶将片段sgrna(ttat)scaffold与载体puc19/hu6-sgrnascaffold连接,产物转化入大肠杆菌dh5α,挑取单克隆菌株,经过培养后经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆为所需要的载体,命名为pu6-sgrna(ttat)scaffold。
(3)构建靶向人tpi1p2基因的pu6-sgrna(ttat)-tpi1p2载体
靶向人tpi1p2基因的sgrna片段由引物退火形成,引物由苏州金唯智公司合成,序列如下(下划线表示粘性末端序列):
正向引物序列为:accgcctagagtggccacagtac;
反向引物序列为:taacgtactgtggccactctagg。
两条引物用超纯水稀释成浓度为10μm的溶液,各取15μl混合后于室温退火3-4小时。获得的片段即为靶向人tpi1p2基因的sgrna片段,其末端带有特定的粘性末端。载体pu6-sgrna(ttat)scaffold经bsai酶切后,经质量浓度为1%的琼脂糖凝胶电泳后回收载体,用t4连接酶将靶向人tpi1p2基因的sgrna片段与经bsai酶切的载体pu6-sgrna(ttat)scaffold连接,产物转化入大肠杆菌dh5α,挑取单克隆菌株,经过培养后经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆为所需要的载体,命名为pu6-sgrna(ttat)-tpi1p2。
(4)构建靶向人emx1基因的pu6-sgrna(ttat)-emx1载体
靶向人emx1基因的sgrna片段由引物退火形成,引物由上海生工公司合成,序列如下(下划线表示粘性末端序列):
正向引物序列为:accgagtccgagcagaagaagaa;
反向引物序列为:taacttcttcttctgctcggact。
两条引物用超纯水稀释成浓度为10μm的溶液,各取15μl混合后于室温退火3-4小时。获得的片段即为靶向人emx1基因的sgrna片段,其末端带有特定的粘性末端。载体pu6-sgrna(ttat)scaffold经bsai酶切后,经质量浓度为1%的琼脂糖凝胶电泳后回收载体,用t4连接酶将靶向人emx1基因的sgrna片段与经bsai酶切的载体pu6-sgrna(ttat)scaffold连接,产物转化入大肠杆菌dh5α,挑取单克隆菌株,经过培养后经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆为所需要的载体,命名为pu6-sgrna(ttat)-emx1。
(5)构建靶向人rankl基因的pu6-sgrna(ttat)-rankl载体
靶向人rankl基因的sgrna片段由引物退火形成,引物由上海生工公司合成,序列如下(下划线表示粘性末端序列):
正向引物序列为:accggcttcttcttcttctctct;
反向引物序列为:taacagagagaagaagaagaagc。
两条引物用超纯水稀释成浓度为10μm的溶液,各取15μl混合后于室温退火3-4小时。获得的片段即为靶向人rankl基因的sgrna片段,其末端带有特定的粘性末端。载体pu6-sgrna(ttat)scaffold经bsai酶切后,经质量浓度为1%的琼脂糖凝胶电泳后回收载体,用t4连接酶将靶向人rankl基因的sgrna片段与经bsai酶切的载体pu6-sgrna(ttat)scaffold连接,产物转化入大肠杆菌dh5α,挑取单克隆菌株,经过培养后经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆为所需要的载体,命名为pu6-sgrna(ttat)-rankl。
以下是发明人给出的具体实施例,需要说明的是,以下的实施例是较佳的例子,本发明并不限于这些实施例。
实施例1:
本实施例构建的表达靶向人tpi1p2基因的高活性sgrna载体pu6-sgrna(ttat)-tpi1p2如下:
高活性sgrna(ttat)scaffold中,将原骨架序列crrnarepeat序列中连续的tttt碱基替换为ttat碱基,并将与其对应互补的trancrna序列中连续aaaa碱基替换为ataa碱基,即改变原sgrna骨架两个碱基(图1)。利用pcr合成该高活性sgrna(ttat)scaffold片段:
上游引物序列seq.id.no.2如下:
gaattcggtctccgttatagagctagaaatagcaagttataataaggctagtccgttat
下游引物序列seq.id.no.3如下:
tactagtaaaaaagcaccgactcggtgccactttttcaagttgataacggactagcctt
5’端包含ecorⅰ且3’端包含speⅰ酶切位点的sgrna(ttat)scaffold骨架的完整序列seq.id.no.1为:
gaattcggtctccgttatagagctagaaatagcaagttataataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttactagt
高活性sgrna骨架载体pu6-sgrna(ttat)scaffold中,u6启动子序列seq.id.no.8如下:
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggac
完整表达元件u6-sgrna(ttat)scaffold部分的序列seq.id.no.4为:
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgggagaccgaattcggtctccgttatagagctagaaatagcaagttataataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttta
在两个bsaⅰ中插入的靶向人tpi1p2基因的sgrna片段的序列seq.id.no.5如下:
cctagagtggccacagtac。
上述的表达靶向人tpi1p2基因的高活性pu6-sgrna(ttat)-tpi1p2的载体构建方法步骤如下:
1、合成将原骨架序列crrnarepeat序列中连续的tttt改为ttat碱基和trancrna序列中与其对应互补的连续aaaa改为ataa碱基的高活性sgrna骨架所需的两条引物,普通pcr获得目标高活性sgrna(ttat)scaffold骨架;
根据已知序列和发明人设计,由上海生工公司合成5’前端带有ecorⅰ位点的上游引物p1如seq.id.no.2,5’前端带有speⅰ位点的下游引物p2如seq.id.no.3。在该上下游引物中,有16bp(互补序列双下划线表示)碱基完全互相匹配(引物如下):
以该引物为模板,使用普通pcr扩增合成sgrna(ttat)scaffold骨架片段
反应体系:
聚合酶链式反应的条件是:98℃15秒,98℃10秒,60℃30秒,72℃10秒,72℃5分钟,25个循环。
2、构建pu6-sgrna(ttat)scaffold载体
将sgrna(ttat)scaffold骨架片段经过ecorⅰ与speⅰ酶切,使用1%的琼脂糖凝胶电泳后回收片段,载体puc19/hu6-sgrnascaffold通过ecorⅰ与speⅰ酶切后,使用1%的琼脂糖凝胶电泳后回收载体;使用t4连接酶将片段sgrna(ttat)scaffold与载体puc19/hu6-sgrnascaffold连接,连接条件为:puc19/hu6-sgrnascaffold载体0.5μl,sgrna(ttat)scaffold片段4.5μl,t4-ligationmix5μl,25℃反应20min后转化感受态细胞dh5α,并涂布于含有100μg/ml的氨苄青霉素的lb平板中。挑取菌落接种到含有100μg/ml的氨苄青霉素的lb培养液中,14小时后,经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆。将所获得阳性克隆命名为pu6-sgrna(ttat)scaffold载体,该pu6-sgrna(ttat)scaffold载体结构图(如图2)所示。
3、构建靶向人tpi1p2基因的pu6-sgrna(ttat)-tpi1p2载体
设计的靶向人tpi1p2基因的sgrna片段由引物退火形成,引物由上海生工公司合成,上游引物p3即seq.id.no.9,下游引物p4即seq.id.no.10(下划线表示粘性末端序列):
p3:accgcctagagtggccacagtac;
p4:taacgtactgtggccactctagg。
两条引物用超纯水稀释成浓度为10μm的溶液,各取15μl混合后于室温退火3-4小时。获得的片段即为靶向人tpi1p2基因的sgrna片段,其末端带有特定的粘性末端。载体pu6-sgrna(ttat)scaffold经bsaⅰ酶切后,经质量浓度为1%的琼脂糖凝胶电泳后回收载体,将靶向人tpi1p2基因的sgrna片段与经bsaⅰ酶切的载体pu6-sgrna(ttat)scaffold连接,连接条件为:puc19/hu6-sgrnascaffold载体0.5μl,靶向人tpi1p2基因的sgrna片段4.5μl,t4-ligationmix缓冲液5μl,25℃反应20min后转化感受态细胞dh5α,并涂布于含有100μg/ml的氨苄青霉素的lb平板中。挑取菌落接种到含有100μg/ml的氨苄青霉素的lb培养液中,14小时后,经碱性裂解法提取质粒dna,通过酶切及测序鉴定获得阳性克隆。将所获得阳性克隆命名为pu6-sgrna(ttat)-tpi1p2。
实施例2:
本实施例构建的表达靶向人emx1基因的高活性sgrna载体pu6-sgrna(ttat)-emx1如下:
实施例1中靶向人tpi1p2基因的sgrna片段由靶向人emx1基因的sgrna片段替换。在两个bsaⅰ中插入的靶向人emx1基因的sgrna片段的序列seq.id.no.6如下:
agtccgagcagaagaagaa。
载体的其它结构与实施例1相同。
其构建方法步骤3中,构建靶向人emx1基因的pu6-sgrna(ttat)-emx1载体的方法是:
靶向人emx1基因的sgrna片段由引物退火形成,引物由上海生工公司合成,上游引物p5即seq.id.no.11,下游引物p6即seq.id.no.12,序列如下(下划线表示粘性末端序列):
p5:accgagtccgagcagaagaagaa;
p6:taacttcttcttctgctcggact。
其余构建方法与实施例1相同,构建成表达靶向人emx1基因的的高活性sgrna载体pu6-sgrna(ttat)-emx1。
实施例3:
本实施例构建的表达靶向人rankl基因的高活性sgrna载体pu6-sgrna(ttat)-rankl如下:
实施例1中靶向人tpi1p2基因的sgrna片段由靶向人rankl基因的sgrna片段替换。在两个bsaⅰ中插入的靶向人rankl基因的sgrna片段的序列seq.id.no.7如下:
gcttcttcttcttctctct。
载体的其它结构与实施例1相同。
其构建方法步骤3中,构建靶向人rankl基因的pu6-sgrna(ttat)-rankl载体的方法是:
靶向人rankl基因的sgrna片段由引物退火形成,引物由上海生工公司合成,上游引物p7即seq.id.no.13,下游引物p8即seq.id.no.14,序列如下(下划线表示粘性末端序列):
p7:accggcttcttcttcttctctct;
p8:taacagagagaagaagaagaagc。
其余构建方法与实施例1相同,构建成表达靶向人rankl基因的的高活性sgrna载体pu6-sgrna(ttat)-rankl。
为了验证构建的高活性sgrna骨架载体有益效果,发明人采用实施例1构建的表达靶向人tpi1p2基因的高活性sgrna载体pu6-sgrna(ttat)-tpi1p2,实施例2构建的表达靶向人emx1基因的高活性sgrna的载体pu6-sgrna(ttat)-emx1,实施例3构建的表达靶向人rankl基因的高活性sgrna的载体pu6-sgrna(ttat)-rankl,并进行了实验,实验情况如下:
1、表达靶向人tpi1p2基因的高活性sgrna的载体对目标基因的编辑效率
将未经修饰的通用型sgrna骨架载体命名为puc19/hu6-sgrnascaffold,在本实验中作为阳性对照。载体puc19/hu6-sgrnascaffold中也插入了靶向人tpi1p2基因的sgrna片段,上游引物为p3即seq.id.no.9,下游引物为p9即seq.id.no.15(aaacgtactgtggccactctagg,仅将引物p4第一个碱基从t变为a)其方法与实施例1中构建方法步骤3相同,获得的载体命名为pu6-sgrna-tpi1p2。接种hek293细胞于6孔板,每孔4×105个细胞,共同转染表达cas9的蛋白的载体hcas9(addgene,#41815)与pu6-sgrna(ttat)-tpi1p2进入细胞;而同时,共同转染表达cas9的蛋白的载体hcas9与pu6-sgrna-tpi1p2进入细胞,作为阳性对照。72小时后提取细胞基因组dna。使用巢式pcr扩增基因组中包含编辑位点的区域。
外巢反应:
上游引物p10即seq.id.no.16,下游引物p11即seq.id.no.17:
p10:aaggctctccgtggaagttg;
p11:agtgcaaaggactagcagca。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,56℃15秒,72℃60秒,72℃5分钟,30个循环。
内巢反应:
上游引物p12即seq.id.no.18,下游引物p13即seq.id.no.19:
p12:gtacactacaaacggcctcct;
p13:agctgtaagcagaatgaagacct。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,56℃15秒,72℃30秒,72℃5分钟,30个循环。
所获得的pcr产物经过pcr仪器变性退火的程序是:
95℃,5分钟;95℃-85℃,每秒降低2℃(-2℃/s);85℃-25℃,每秒降低0.1℃(-0.1℃/s)。
pcr变性退火产物经过质量浓度为1%的琼脂糖凝胶电泳后回收后,测量含量,然后使用经典的t7endonucleaseⅰ内切酶检测法检测各组中目标基因的编辑效率。具体方法如下:每组取pcr产物dna500ng,0.5μl的t7endonucleaseⅰ(neb,m0302s)内切酶,2μl的10×buffer,补水至总体积20μl。37℃,反应25分钟。结束后进行1.5%的琼脂糖凝胶电泳检测。t7endonucleaseⅰ内切酶能够将pcr产物中不配对的部分切断,如果pcr产物被切开成两条小的条带,则说明其在目标位置发生了基因组编辑,产生了碱基突变,而两条小的条带的亮度越亮,则说明基因组编辑的效率越高。结果见图4所示,在相同的转染条件下,与control组(阴性对照,空转cas9质粒细胞)相比,pu6-sgrna-tpi1p2与pu6-sgrna(ttat)-tpi1p2均能介导cas9进行基因组编辑,但载体pu6-sgrna(ttat)-tpi1p2介导cas9进行基因组编辑的效应强于pu6-sgrna-tpi1p2。此结果说明,经过碱基改变以后的高活性sgrna表达载体介导cas9对人tpi1p2位点基因组编辑的效应高于普通sgrna载体。
2、表达靶向人emx1基因的高活性sgrna的载体对目标基因的编辑效率
发明人将在验证实验1中使用的载体puc19/hu6-sgrnascaffold中也插入了靶向人emx1基因的sgrna序列,上游引物为p5即seq.id.no.11,下游引物为p14即seq.id.no.20(aaacttcttcttctgctcggact,仅将引物p6的第一个碱基从t变为a),其方法与实施例1中构建方法步骤3相同,获得的载体命名为pu6-sgrna-emx1。接种hek293细胞(同实验1)后并转染表达cas9的蛋白的载体hcas9(addgene,#41815)与pu6-sgrna(ttat)-emx1进入细胞;以及共同转染表达cas9的蛋白的载体hcas9与pu6-sgrna-emx1进入细胞,作为阳性对照。72小时后提取细胞基因组dna并使用巢式pcr扩增基因组中包含编辑位点的区域。
外巢反应:
上游引物p15即seq.id.no.21,下游引物p16即seq.id.no.22:
p15:ggagcagctggtcagagggg;
p16:gggaagggggacactgggga。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,60℃15秒,72℃42秒,72℃5分钟,30个循环。
内巢反应:
上游引物p16即seq.id.no.23,下游引物p17即seq.id.no.24:
p17:gcctgagtgttgaggccc;
p18:gaaggccaagtggtcccag。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,60℃15秒,72℃42秒,72℃5分钟,30个循环。
所获得的pcr产物经过pcr变性退火并进行t7eⅰ酶切,具体操作同实验1。结果见图5所示,虽然相同的转染条件下,pu6-sgrna-emx1与pu6-sgrna(ttat)-emx1均能介导cas9进行基因组编辑,但是载体pu6-sgrna(ttat)-emx1介导cas9进行基因组编辑的效应强于pu6-sgrna-emx1。此结果说明,经过碱基改变以后的高活性sgrna表达载体介导cas9对人emx1位点基因组编辑的效应高于普通sgrna载体。
3、表达靶向人rankl基因的高活性sgrna的载体对目标基因的编辑效率
发明人将在验证实验1中使用的载体puc19/hu6-sgrnascaffold中也插入了靶向人rankl基因的sgrna序列,上游引物为p7即seq.id.no.13,下游引物将p19即seq.id.no.25(taacagagagaagaagaagaagc,仅将引物p8中的第一个碱基从t变为a),其方法与实施例1中构建方法步骤3相同,获得的载体命名为pu6-sgrna-rnakl。接种hek293细胞(同实验1)后并转染表达cas9的蛋白的载体hcas9(addgene,#41815)与pu6-sgrna(ttat)-rankl进入细胞;以及共同转染表达cas9的蛋白的载体hcas9与pu6-sgrna-rankl进入细胞,作为阳性对照。72小时后提取细胞基因组dna并使用巢式pcr扩增基因组中包含编辑位点的区域。
外巢反应:
上游引物p20即seq.id.no.26,下游引物p21即seq.id.no.27:
p20:ttgctagggcaacaagtgga;
p21:tggcctctggatctcaccaa。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,62℃15秒,72℃2分钟,72℃5分钟,30个循环。
内巢反应:
上游引物p22即seq.id.no.28,下游引物p23即seq.id.no.29:
p22:tcacccaatgcagagctgaag;
p23:tctctgatgtttgtgggggaag。
反应体系:
聚合酶链式反应的条件是:95℃3分钟,95℃15秒,60℃15秒,72℃25秒,72℃5分钟,30个循环。
所获得的pcr产物经过pcr变性退火并进行t7eⅰ酶切,具体操作同实验1。结果见图6所示,虽然相同的转染条件下,pu6-sgrna-rankl与pu6-sgrna(ttat)-rankl均能介导cas9进行基因组编辑,但是载体pu6-sgrna(ttat)-rankl介导cas9进行基因组编辑的效应强于pu6-sgrna-rankl。此结果说明,经过碱基改变以后的高活性sgrna表达载体介导cas9对人rankl位点基因组编辑的效应高于普通sgrna载体。
上述实验1、实验2和实验3说明,高活性sgrna的载体介导cas9对目标位点基因组编辑的效应显著强于普通sgrna载体。
序列表
<110>西安交通大学
<120>一种双碱基突变的高活性sgrna骨架、sgrna骨架载体及其应用
<160>29
<170>siposequencelisting1.0
<210>1
<211>101
<212>dna
<213>人工序列(artificialsequence)
<400>1
gaattcggtctccgttatagagctagaaatagcaagttataataaggctagtccgttatc60
aacttgaaaaagtggcaccgagtcggtgcttttttactagt101
<210>2
<211>59
<212>dna
<213>人工序列(artificialsequence)
<400>2
gaattcggtctccgttatagagctagaaatagcaagttataataaggctagtccgttat59
<210>3
<211>59
<212>dna
<213>人工序列(artificialsequence)
<400>3
tactagtaaaaaagcaccgactcggtgccactttttcaagttgataacggactagcctt59
<210>4
<211>353
<212>dna
<213>人工序列(artificialsequence)
<400>4
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagag60
ataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtaga120
aagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcat180
atgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaagga240
cgaaacaccgggagaccgaattcggtctccgttatagagctagaaatagcaagttataat300
aaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttta353
<210>5
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>5
cctagagtggccacagtac19
<210>6
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>6
agtccgagcagaagaagaa19
<210>7
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>7
gcttcttcttcttctctct19
<210>8
<211>241
<212>dna
<213>人工序列(artificialsequence)
<400>8
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagag60
ataattagaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtaga120
aagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcat180
atgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaagga240
c241
<210>9
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>9
accgcctagagtggccacagtac23
<210>10
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>10
taacgtactgtggccactctagg23
<210>11
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>11
accgagtccgagcagaagaagaa23
<210>12
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>12
taacttcttcttctgctcggact23
<210>13
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>13
accggcttcttcttcttctctct23
<210>14
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>14
taacagagagaagaagaagaagc23
<210>15
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>15
aaacgtactgtggccactctagg23
<210>16
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>16
aaggctctccgtggaagttg20
<210>17
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>17
agtgcaaaggactagcagca20
<210>18
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>18
gtacactacaaacggcctcct21
<210>19
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>19
agctgtaagcagaatgaagacct23
<210>20
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>20
aaacttcttcttctgctcggact23
<210>21
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>21
ggagcagctggtcagagggg20
<210>22
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>22
gggaagggggacactgggga20
<210>23
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>23
gcctgagtgttgaggccc18
<210>24
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>24
gaaggccaagtggtcccag19
<210>25
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>25
taacagagagaagaagaagaagc23
<210>26
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>26
ttgctagggcaacaagtgga20
<210>27
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>27
tggcctctggatctcaccaa20
<210>28
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>28
tcacccaatgcagagctgaag21
<210>29
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>29
tctctgatgtttgtgggggaag22
- 还没有人留言评论。精彩留言会获得点赞!