一种吲哚二酮哌嗪类生物碱及其制备方法和用途与流程
本发明属于生物医药领域,具体涉及一种吲哚二酮哌嗪类生物碱及其制备方法和用途,包括菌株发酵,分离纯化过程,结构确证,以及对白血病细胞株抑制活性作用等。
背景技术:
吲哚二酮哌嗪生物碱及其衍生物主要来自微生物的代谢产物,它们通常从真菌中分离得到,例如青霉属(penicillium)、曲霉属(aspergillus)、拟盘多毛孢属(pestalotiopsis)等。其结构特点是具有一定的缩合产物特征,包括完整的l-色氨酸和其他氨基酸,如l-脯氨酸,l-苯丙氨酸,l-组氨酸或l-亮氨酸等,从而形成吲哚二酮哌嗪单元。吲哚二酮哌嗪生物碱及其衍生物具有广泛的生物活性,如抗癌、抗菌、抗病毒、抗氧化等。因此,它们具有用于药物或作为药物开发的潜力。(ma,y.m.structuraldiversityandbiologicalactivitiesofindolediketopiperazinealkaloidsfromfungi.journalofagriculturalandfoodchemistry.2016,64(35):6659-6671)
随着科研人员致力于研究和开发新的活性药物,吲哚二酮哌嗪及其衍生物因其广泛的生物活性越来越受到科学界的关注。本专利的重点是发明人在对木竹子内生真菌的持续研究中,发现了一株内生曲霉菌(aspergillussp.gzwmjz-258),在对此菌株次级代谢产物的研究过程中,通过分离鉴定得到两个新颖的吲哚二酮哌嗪生物碱,具有罕见的6/6/5/5/6/5/5七环骨架,并有一定的抑制白血病细胞株的活性。迄今为止,现有技术中未见有此种七环骨架的吲哚二酮哌嗪生物碱及其活性的研究报道。
技术实现要素:
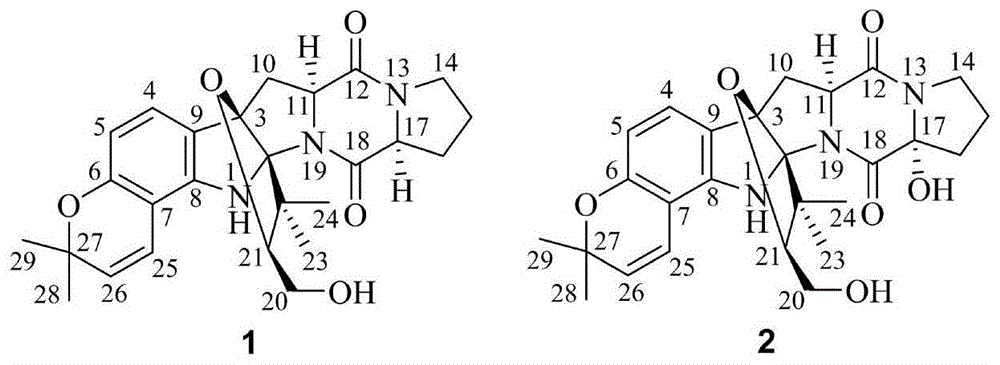
本发明的目的在于从木竹子内生真菌发酵产物中提取、分离和纯化生物碱具有对白血病细胞株mv4-11具有选择性抑制作用,可在抗白血病药物中得到应用。提供一种吲哚二酮哌嗪类生物碱及其制备方法和用途。本发明的目的及解决其主要技术问题是采用以下技术方案来实现的:一种吲哚二酮哌嗪类生物碱,该生物碱从木竹子内生真菌的发酵产物中分离纯化得到,其结构式为式(1)和式(2):
其中所述的内生真菌命名为:aspergillussp.gzwmjz-258,于2019年4月28日保藏单位于中国典型培养物保藏中心,地址中国武汉,保藏号为cctccno:m2019305。
所述的化合物1和2的制备方法,包括以下步骤:
a菌种准备:使用的纯化培养基重量份数为马铃薯200-300份,葡萄糖20-25份,琼脂15-20份,加入到1l纯净水中,调节ph值6.5~7.5,制成斜面,至于常温状态下,接种aspergillussp.gzwmjz-258的菌丝体,于28-35℃下培养3天,作为菌种;
b发酵种子液制备:按重量份数为:麦芽糖10-20份,谷氨酸钠5-15g,磷酸二氢钾0.5-1份,硫酸镁0.3-1份,葡萄糖10-20份,酵母膏3-5份,玉米浆1-2份,甘露醇20-30份,加入到1l纯净水中,调节ph值6.5~7.5,培养温度28-35℃,在500ml锥形瓶中分别装入150ml,接种步骤a得到的菌种,180r·min-1下震荡培养2天,将该培养物作为发酵种子液;
c接种:采用固体发酵方式,每个发酵袋装入大米50-100份、纯净水50-100份,接种步骤b得到的发酵种子液,每袋的接种量为10ml,28-35℃下静置培养30-35天后得到发酵培养基;
d萃取:用乙酸乙酯浸泡步骤c所述的发酵培养基48-72小时,再用搅拌器搅拌提取3-5次,每次30-40分钟,将上清液合并后于旋转蒸发仪回收乙酸乙酯,得到粗浸膏;
e单体分离纯化:将步骤c所得粗浸膏经柱层析分离,得到新结构为式(1)和式(2)的生物碱;所述层析分离方法包括硅胶柱层析法,凝胶柱层析法,半制备高效液相色谱法。
发明人在对木竹子内生真菌的持续研究中,发现了一株曲霉属内生真菌(aspergillussp.gzwmjz-258),其特征在于,所述菌株的命名为:aspergillussp.gzwmjz-258,于2019年4月28日保藏单位于中国典型培养物保藏中心,地址中国武汉,保藏号为cctccno:m2019305。发明人对其发酵产物在提取、分离和纯化方面进行了系统的研究,得到了2个新骨架吲哚二酮哌嗪生物碱1和2。制备的化合物1和2具有罕见的6/6/5/5/6/5/5七环骨架,这在吲哚二酮哌嗪类生物碱中首次报道;对白血病细胞株mv4-11具有一定的选择性抑制作用,可在抗白血病药物中得到应用。本发明与现有技术相比具有明显的优点和有益效果。由以上技术方案可知,本发明化合物对flt3-itd突变的急性骨髓性细胞株mv4-11具有一定的抑制作用,但是对其他几种肿瘤细胞株,如a549,k562以及hl-60的抑制活性较弱;可被开发为高效低毒的预防和治疗白血病的药物。本发明综合应用质谱、核磁共振氢谱、核磁共振碳谱、以及核磁共振二维谱等分析技术对发酵产物中的化合物1和2进行结构鉴定。
附图说明
图1是本发明化合物1和2的结构式
图2是本发明化合物1的高分辨质谱图
图3是本发明化合物2的高分辨质谱图
图4是本发明化合物1的核磁共振氢谱图
图5是本发明化合物2的核磁共振氢谱图
图6是本发明化合物1的核磁共振碳谱图
图7是本发明化合物2的核磁共振碳谱图
具体实施方式
实施例1:
(一)化合物1和2的制备
(1)培养基准备
使用的纯化培养基成分为:马铃薯200份,葡萄糖20份,琼脂20份,纯净水1l。高温灭菌,制成斜面,置于常温状态下,无杂菌生长时,接种内生真菌aspergillussp.gzwmjz-258的菌丝体,于28℃下静置培养。
(2)发酵种子液制备
在500ml锥形瓶中分别装入150ml种子液培养基,所述种子液培养基成分为:麦芽糖20份,谷氨酸钠10份,磷酸二氢钾0.5份,硫酸镁0.3份,葡萄糖10份,酵母膏3份,玉米浆1份,甘露醇20份,加入到1l纯净水中,调节ph值6.5~7.5,121℃,30min高温灭菌后进行接种,28℃下,180r·min-1下震荡培养2天,将该培养物作为种子液;
(3)接种
采用固体发酵方式,准备100个发酵袋,每个发酵袋装入大米50份、纯净水60份,121℃,30min高温灭菌后,接种上述种子液培养,每袋的接种量为10ml,28℃下静置培养30天。
(4)萃取
将上述发酵得到的发酵液及菌丝体用乙酸乙酯浸泡48小时,然后分批次用搅拌器搅拌提取,每批次提取三次,每次提取30分钟。静置后合并上清液于旋转蒸发仪中回收乙酸乙酯,得到粗浸膏67克。
(5)单体分离纯化
二氯甲烷:甲醇(1:1)溶解粗浸膏后用100-200目硅胶拌样进行硅胶柱层析,用二氯甲烷:甲醇梯度洗脱(100:1–2:1),tlc检测,合并相同的组分,共得到11个组分。组分9再经过硅胶柱层析,石油醚:乙酸乙酯梯度洗脱(100:1-1:1),tlc检测,合并相同组分,共得到15个组分。将组分9-12经过凝胶柱层析(甲醇柱),tlc检测,合并相同的组分,共得到9个组分,将9-15-6组分用半制备高效液相色谱法分离(50%甲醇/水),得到化合物1(5.0mg)和2(8.8mg)。
(二)化合物1和2的结构鉴定
通过高分辨质谱,紫外光谱,红外光谱,旋光,核磁共振等数据进行综合分析,从而确定化合物1和2的结构,其理化性质如下:
化合物1:白色粉末;分子式c26h31n3o5;分子量465;uv(meoh)λmax(logε)210(2.54),236(2.72),289(0.90)nm;ir(kbr)vmax3427,2972,1664,1610,1461,1413,1325,1157,1063,733cm-1;hresimsm/z488.2161[m+na]+;[α]d20-168(c0.1,meoh),其核磁共振氢谱、核磁共振碳谱数据如表1所示。
化合物2:白色粉末;分子式c26h31n3o6;分子量481;uv(meoh)λmax(logε)203(4.68),213(2.83),237(3.07),287(0.88)nm;ir(kbr)νmax3419,2976,1664,1610,1461,1423,1325,1153,1063,1022,733cm-1;hresimsm/z504.2107[m+na]+;[α]d20-192(c0.1,meoh),其核磁共振氢谱、核磁共振碳谱数据如表1所示。
表1.化合物1和2的核磁共振氢谱、碳谱数据
实施例2
为了进一步验证本发明合成的化合物的有益效果,通过对化合物1和2进行肿瘤细胞抑制活性测试,具体实验如下:
被测样品溶液的配制:测试样品为上述实施例1中分离得到的化合物1和2。准确称取适量样品,用dmso配制成所需浓度的溶液,供活性测试。
细胞系及细胞的继代培养:活性测试采用mv-4-11、k562、hl-60和a549细胞。各种细胞均用含10%fbs的rpmi-1640培养基,在37℃通入5%二氧化碳的培养箱中继代培养。
本实验采用celltiterglo(ctg)法检测不同药物对mv-4-11、k562、hl-60和a549细胞的生长抑制作用。活性测试时,取对数生长期的mv-4-11、k562、hl-60和a549细胞,用新鲜的imdm培养基配制成密度为每毫升2×104个细胞的细胞悬液,细胞经计数后接种于96孔培养板,2000细胞/100μl/孔,每孔加入90μl培养液(含血清)培养过夜,再加入10μl药物溶液,继续培养48小时。然后加入100μlctg,室温静置10min,测定发光值,0.5ms。按照下式计算每个浓度下的细胞增殖率(%):ir%=(对照组化学发光值-实验组化学发光值)/对照组化学发光值×100%。应用graphpad软件,计算ic50;实验结果如表2所示。
表2.化合物1和2对4种肿瘤细胞的增殖抑制活性(ic50)
这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修饰同样落入本发明权利要求所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!