一种二氢嘧啶类前药及其制备方法和应用与流程
本发明属于医药技术领域,具体涉及一种二氢嘧啶类前药及其制备方法与制药用途。
背景技术:
乙型病毒性肝炎(viralhepatitistypeb),简称乙肝(hepatitisb),是由乙型肝炎病毒(hepatitisbvirus,hbv)所致的重大传染性疾病,长期发展可导致急慢性病毒性肝炎、肝硬化和原发性肝细胞癌(hepatocellularcarcinoma,hcc)等一系列并发症。据世界卫生组织(worldhealthorganization,who)统计,全球近20亿人感染过hbv,其中约2.6亿人为慢性hbv感染者,平均每年约有60万人死于hbv感染所致的相关疾病。截止2019年,美国食品药品监督管理局(u.s.foodanddrugadministration,fda)批准用于预防和治疗慢性乙型肝炎(chronichepatitisb,chb)的药物主要分为干扰素和dna聚合酶抑制剂两大类,上市药物对于乙肝患者起到一定的治疗作用,但仍存在缺陷。干扰素通过皮下注射给药,耐受性差,同时治愈率低,带来抑郁和关节痛等副作用,不适合孕妇、有自身免疫性疾病和晚期肝硬化的患者使用;dna聚合酶抑制剂疗程长,无法清除cccdna,易产生耐药性,停药后复发率高。因此研发新型高效、低毒和抗耐药性的非核苷类乙肝病毒抑制剂具有重要意义。
核心蛋白是hbv核壳体组成的主要结构蛋白,在病毒进化过程中相对保守,并且核心蛋白的组装在乙肝病毒生命周期中发挥着重要作用。然而,目前还没有相关靶点的药物上市。二氢嘧啶(heteroaryldihydropyrimidines,hap)类hbv核衣壳蛋白抑制剂gls4具有较好的抗病毒活性,其抑制hbvdna复制活性已达到纳摩尔级别(hepg2.2.15细胞系,ic50=12nm),对阿德福韦耐药株也有较好的活性(rta181v,ic50=0.161μm;rtn236t,ic50=0.131μm),具有良好的开发前景,目前正由广东东阳光药业有限公司进行ii期临床研究。但是gls4的水溶性较差,口服生物利用度不高(clogp=4.7,f=14%),限制了其进一步开发。针对gls4存在的缺陷,本发明通过前药的策略,设计合成了一类含“5-甲基-2-氧代-1,3-二氧杂环戊烯”基团的二氢嘧啶类前药,此类化合物在现有技术中未见相关报道。
技术实现要素:
针对现有技术的不足,本发明提供了一种二氢嘧啶类前药及其制备方法,本发明还提供了上述化合物作为非核苷类hbv抑制剂的活性筛选结果及其应用。
本发明的技术方案如下:
一、二氢嘧啶类前药
本发明涉及的二氢嘧啶类前药,具有如下i-5所示的结构:
二、二氢嘧啶类前药的制备方法
本发明二氢嘧啶类前药的合成路线以2-溴-4-氟苯甲醛、2-噻唑甲脒盐酸盐(i-1)和乙酰乙酸乙酯为原料,通过“biginelli”反应环合得到中间体i-2;i-2在四氯化碳中与n-溴代丁二酰亚胺发生取代反应得到中间体i-3;i-3在dmf溶液中与吗啉发生取代反应得到中间体i-4;再通过一步烃化反应得到目标化合物i-5:
合成路线如下:
试剂及条件:(i)2-溴-4-氟苯甲醛,乙酰乙酸乙酯,醋酸钠,无水乙醇,80℃;(ii)n-溴代丁二酰亚胺,四氯化碳,50℃;(iii)吗啉,n,n-二甲基甲酰胺,25℃;(iv)4-溴甲基-5-甲基-1,3-间二氧杂环戊烯-2-酮,氢化钠,n,n-二甲基甲酰胺,25℃。
根据本发明优选的,二氢嘧啶类前药的制备方法,步骤如下:
(1)室温条件下将3.05mmol2-噻唑甲脒盐酸盐溶于50ml无水乙醇中,依次加入乙酰乙酸乙酯4.60mmol,2-溴-4-氟苯甲醛4.60mmol和醋酸钠6.13mmol,80℃下加热反应6h;反应结束后将反应液取出冷却至室温,减压除去无水乙醇,加入水60ml,用乙酸乙酯萃取3次,每次25ml,收集并合并有机相,用25ml饱和氯化钠洗一次,再用无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,采用二氯甲烷-正己烷体系重结晶得到化合物i-2;
(2)将1.17mmol中间体i-2溶于50ml四氯化碳中,室温条件下少量多次加入1.24mmolnbs,室温下反应2h;反应结束后,减压除去四氯化碳,加入水50ml,用乙酸乙酯萃取3次,每次20ml,收集并合并有机相,用25ml饱和氯化钠洗一次,再用无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,采用二氯甲烷-正己烷体系重结晶得到化合物i-3;
(3)将1.00mmol中间体i-3溶于50mldmf中,室温条件下加入2.00mmol吗啉,室温搅拌过夜;反应结束后,加入100ml水,用乙酸乙酯萃取3次,每次20ml,收集并合并有机相,用25ml饱和氯化钠洗一次,无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,用二氯甲烷-正己烷体系重结晶得到化合物i-4;
(4)将0.39mmol中间体i-4溶于2ml超干dmf中,冰浴条件下分批加入0.47mmolnah,室温搅拌1h;再加入0.47mmol4-溴甲基-5-甲基-1,3-间二氧杂环戊烯-2-酮,套上干燥管,室温搅拌过夜;反应结束后,加入50ml水,乙酸乙酯萃取3次,每次20ml,合并有机相,25ml饱和食盐水洗一次,无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,干燥后得黄色油状物i-5。
三、二氢嘧啶类前药的应用
本发明公开了二氢嘧啶类前药抗hbv活性筛选结果、初步成药性评价及其作为抗hbv抑制剂的应用。通过实验证明本发明的二氢嘧啶类前药可作为经典的hbv非核苷类抑制剂应用。
如表1所示,对所合成的目标化合物i-5进行了体外细胞水平的抗hbv活性评价,通过cck-8法测定了i-5的细胞毒性;通过pcr法测定了i-5的hbvdna复制抑制活性;通过elisa法测定了i-5的抗原分泌抑制活性。选择先导化合物gls4和上市药物拉米夫定为阳性对照,每个化合物设置五个浓度梯度(50μm、5μm、0.5μm、0.05μm和0.005μm),分别计算出半数抑制浓度cc50、ic50和选择性系数si。
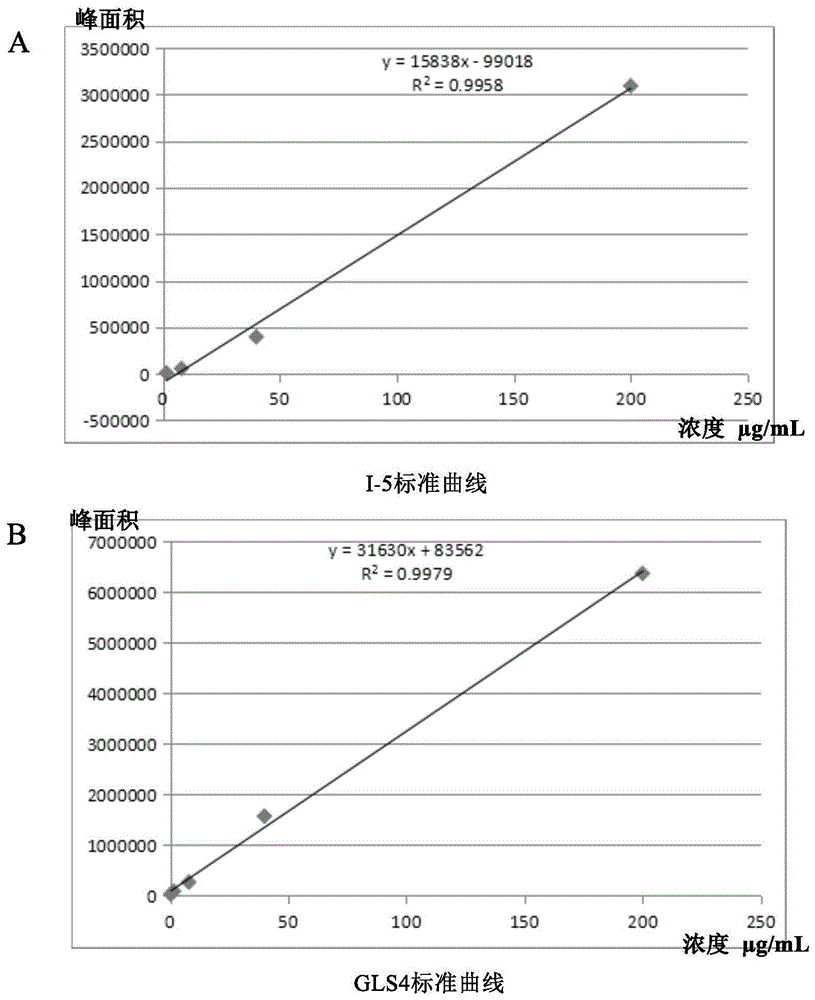
如图1和表2所示,对i-5进行了水溶性的测试,通过高效液相色谱法建立其标准曲线,再将一定量i-5的饱和甲醇溶液加到水和不同ph的pbs缓冲液中,测定吸收峰面积,代入标准曲线计算其在不同溶液中的饱和溶解度。
如图2所示,对i-5进行了小鼠体内的急性毒性实验,以最大剂量(2g/kg)灌胃后,一周内每天监测小鼠是否死亡,记录其体重增长情况和个体行为(嗜睡、阵挛性惊厥、厌食、皱皮等有无异常)。
如表3、表4、图3所示,对i-5进行了大鼠体内的药代动力学实验,分别对大鼠单次静脉注射(1mg/kg)、灌胃(10mg/kg)给予化合物i-5,于不同时间点采集血液,测定大鼠血浆中前药i-5和原药gls4的浓度,并计算相关药代动力学参数。
本发明的二氢嘧啶类前药是一类结构新颖的非核苷类hbv抑制剂,可作为抗hbv的先导化合物。
本发明的二氢嘧啶类前药可作为非核苷类hbv抑制剂应用。具体地说,作为hbv抑制剂用来制备抗乙肝药物。
一种抗hbv药物组合物,包括本发明的二氢嘧啶类前药和一种或多种药学上可接受载体或赋形剂。
本发明公开了二氢嘧啶类前药、其制备方法、抗hbv活性筛选结果、初步成药性评价及其作为抗hbv抑制剂的首次应用。实验证明本发明的二氢嘧啶类前药可作为hbv抑制剂用于制备抗乙肝药物。
附图说明
图1是i-5和gls4的标准曲线图;
图2是i-5的急性毒性试验结果图;
图3是i-5口服和注射平均血药浓度-时间曲线。
具体实施方式
下面结合实施例对本发明做进一步说明,但不限于此。
合成路线:
试剂及条件:(i)2-溴-4-氟苯甲醛,乙酰乙酸乙酯,醋酸钠,无水乙醇,80℃;(ii)n-溴代丁二酰亚胺,四氯化碳,50℃;(iii)吗啉,n,n-二甲基甲酰胺,25℃;(iv)4-溴甲基-5-甲基-1,3-间二氧杂环戊烯-2-酮,氢化钠,n,n-二甲基甲酰胺,25℃。
实施例1.化合物i-2的制备
室温条件下将2-噻唑甲脒盐酸盐(0.50g,3.05mmol)溶于无水乙醇(50ml)中,依次加入乙酰乙酸乙酯(600μl,4.60mmol),2-溴-4-氟苯甲醛(0.93g,4.60mmol)和醋酸钠(0.50g,6.13mmol),80℃下加热反应6h;反应结束后将反应液取出冷却至室温,减压除去无水乙醇,加入水(60ml),用乙酸乙酯萃取(25ml×3),收集并合并有机相,用饱和氯化钠洗一次(25ml),再用无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,采用二氯甲烷-正己烷体系重结晶得到黄色粉末0.75g,产率:58%;熔点158-160℃;1hnmr(400mhz,dmso-d6)δ9.92(s,1h),7.97(d,j=2.8hz,1h),7.89(s,1h),7.59–7.50(m,1h),7.42–7.31(m,1h),7.23(t,j=8.3hz,1h),5.98(s,1h),3.94(q,j=6.9hz,2h),2.48(s,3h),1.03(t,j=7.0hz,3h);13cnmr(100mhz,dmso-d6)δ166.07,163.13,159.93,147.99,144.78,143.69,141.19,131.14(d,j=8.7hz),124.85,122.94(d,j=9.6hz),119.98(d,j=24.2hz),115.85(d,j=21.0hz),97.33,59.57,58.14,17.86,14.46;ei-ms:426.04[m+2+h]+.
实施例2.化合物i-3的制备
将中间体i-2(0.50g,1.17mmol)溶于四氯化碳(50ml)中,室温条件下少量多次加入nbs(0.22g,1.24mmol),室温下反应2h;反应结束后,减压除去四氯化碳,加入水(50ml),用乙酸乙酯萃取(20ml×3),收集并合并有机相,用饱和氯化钠洗一次(25ml),再用无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,采用二氯甲烷-正己烷体系重结晶得到黄色粉末0.35g;产率:59%;熔点125-127℃;1hnmr(400mhz,cdcl3)δ7.83(d,j=2.4hz,1h),7.64–7.34(m,3h),7.32(d,j=7.5hz,1h),7.01(d,j=7.0hz,1h),6.12(d,j=38.9hz,1h),4.93(d,j=8.1hz,1h),4.59(d,j=8.1hz,1h),4.12(d,j=6.8hz,2h),1.16(t,j=7.0hz,3h).13cnmr(100mhz,cdcl3)δ164.73,163.27,162.04,160.76,155.66,150.28,143.87,143.01,137.84,130.60(d,j=8.6hz),124.62,123.45,122.10(d,j=9.2hz),120.26(d,j=24.8hz),115.72(d,j=20.9hz),106.39,60.72,51.61,31.79,14.03;ei-ms:499.90[m-h]-,501.94[m+2-h]-,503.91[m+4-h]-.
实施例3.化合物i-4的制备
将中间体i-3(0.50g,1.00mmol)溶于dmf(50ml)中,室温条件下加入吗啉(173μl,2.00mmol),室温搅拌过夜;反应结束后,加入大量水(100ml),用乙酸乙酯萃取(20ml×3),收集并合并有机相,用饱和氯化钠洗一次(25ml),无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,用二氯甲烷-正己烷体系重结晶得到黄色粉末0.40g,产率:79%;熔点124-127℃;1hnmr(400mhz,dmso-d6)δ9.69(s,1h),8.04(d,j=3.1hz,1h),7.95(d,j=2.5hz,1h),7.57(dd,j=8.5,1.9hz,1h),7.40(dd,j=8.6,6.2hz,1h),7.22(td,j=8.5,2.4hz,1h),6.04(s,1h),4.03–3.85(m,4h),3.68(t,j=4.2hz,4h),2.55(t,j=7.2hz,4h),1.06(t,j=7.1hz,3h);13cnmr(100mhz,dmso-d6)δ165.64,162.49,160.02,146.81,144.36,144.08,140.67(d,j=3.3hz),131.38(d,j=8.7hz),125.20,123.05(d,j=9.8hz),120.06(d,j=24.3hz),115.96(d,j=20.8hz),97.65,66.89,59.86,58.65,56.37,53.66,14.46;ei-ms:509.15[m+h]+.
实施例4.化合物i-5的制备
将中间体i-4(0.20g,0.39mmol)溶于2ml超干dmf中,冰浴条件下分批加入nah(11.3mg,0.47mmol),室温搅拌1h;再加入4-溴甲基-5-甲基-1,3-间二氧杂环戊烯-2-酮(67μl,0.47mmol),套上干燥管,室温搅拌过夜;反应结束后,加入50ml水,乙酸乙酯萃取三次(20ml×3),合并有机相,饱和食盐水洗一次(25ml),无水硫酸钠干燥;旋蒸除去溶剂,拌样,硅胶柱分离,干燥后得黄色油状物0.05g,产率:20%;1hnmr(400mhz,dmso-d6)δ8.02(q,j=3.3hz,2h,thiazole-h),7.62(dd,j=8.5,2.5hz,1h,ph-h),7.41(dd,j=8.7,6.1hz,1h,ph-h),7.28(td,j=8.5,2.5hz,1h,ph-h),5.93(s,1h,dihydropyrimidine-h),5.72(d,j=16.6hz,1h,dihydropyrimidine-ch2),4.82(d,j=16.6hz,1h,dihydropyrimidine-ch2),4.10–4.01(m,2h,ch2ch3),3.86(d,j=13.0hz,1h,morpholine-ch2),3.55(t,j=4.3hz,4h,morpholine-h),3.49(d,j=13.0hz,1h,morpholine-ch2),2.52(d,j=10.4hz,4h,morpholine-h),2.03(s,3h,ch3),1.14(t,j=7.1hz,3h,ch2ch3);13cnmr(100mhz,dmso-d6)δ165.31,163.97,160.72,152.61,152.09,149.89,144.87,138.87,137.89(d,j=3.3hz),133.92,131.85(d,j=9.1hz),126.81,122.20(d,j=9.9hz),120.25(d,j=24.7hz),116.71(d,j=21.1hz),109.79,66.91,60.64,59.20,58.30,53.76,44.03,14.51,9.60;ei-ms:623.3[m+2+h]+.
实施例5.化合物i-5的体外抗hbv活性评价
测试原理:hbv转染的肝癌细胞hepg2.2.15细胞株,在进行细胞培养时能够分泌hbv病毒颗粒(包含病毒dna和抗原)。在抗hbv目标化合物的干预下,细胞产生的dna和抗原会有所变化,因此检测细胞产生的hbvdna和抗原,参照未加药对照组的含量,可以反映样品药物的抗病毒活性作用。以gls4和拉米夫定为阳性对照药,用聚合酶链反应(pcr)检测药物抑制hbvdna复制量的50%时的浓度数值ic50;通过elisa法检测药物抑制抗原分泌量的50%时的浓度数值ic50;运用cck-8检测样品药物导致50%细胞毒性死亡的数值浓度为cc50值;并计算出待测化合物的“选择性系数”(si,selectivityindex),计算公式:si=cc50/ic50。
测试方法:(1)细胞毒性实验。配成实验所需样品储备浓度(20mmol/l),用hepg2.2.15细胞培养液配制5个稀释浓度(50μmol/l和5μmol/l、0.5μmol/l、0.05μmol/l、0.005μmol/l),设立空白对照并以gls4和拉米夫定作为的阳性对照药。加入96孔板细胞培养板,每浓度设置3个复孔,每4天换同浓度药液并设无药细胞对照组,共培养9天。用cck-8法检测细胞存活率,确定药物对hepg2.2.15细胞的毒性。
(2)抑制hbvdna合成实验。hepg2.2.15细胞在96孔细胞培养板中培养24小时后,加入含药培养液,继续培养8天(每4天换液一次),收集上清液,用探针法进行pcr检测。
(3)抑制抗原分泌实验。hepg2.2.15细胞在96孔细胞培养板中培养24小时后,加入含药培养液,继续培养8天(每4天换液一次),收集上清液,用elisa法检测抗原分泌量。
表1目标化合物i-5、先导化合物gls4和上市药物拉米夫定(3-tc)的抗hbv活性
如表1所示,i-5的细胞毒性较低(cc50>50μm),表现了较好的抑制hbvdna复制活性,其ic50为0.041±0.045μm,与先导化合物gls4相当(0.035±0.025μm),是上市药物拉米夫定的40多倍(1.73±0.28μm)。除此之外,i-5表现出了较好的hbsag和hbeag分泌抑制活性,其ic50分别为17.87±0.8μm和0.39±0.1μm,选择性系数分别大于2.8和128.8,与先导化合物gls4水平相当(ic50分别为15.82±0.4μm和0.18±0.03μm),远高于拉米夫定(ic50>50μm)。
实施例6.化合物i-5的水溶性测试
(1)流动相预处理。实验开始前将所用的流动相(色谱甲醇和蒸馏水)微开瓶盖,超声除气约3h。
(2)预测待测化合物的溶解度。用dmso溶解待测化合物,配成10mg/ml的母液。取10μl母液加到1ml纯净水中,在漩涡混合器上充分振荡,平衡后肉眼观察溶液中是否有悬浮物。如有,说明已饱和,可进行后续操作;若无,则应继续增大母液的浓度。
(3)标准曲线的制作。将步骤(2)中的母液在ep管中等倍稀释成200μg/ml、40μg/ml、8μg/ml、1.6μg/ml和0.32μg/ml的标曲溶液,有机膜过滤后进样,测定不同浓度c下的峰面积a,以c和a为横纵坐标建立标准曲线,并计算得到标准曲线方程a=kc+b。
(4)待测溶液的配制。取10μl步骤(2)中的母液加到1ml纯净水或1mlpbs缓冲液中,充分振荡混匀(30min),静置备用。(纯净水和pbs缓冲液应先用孔径0.22μm的绿色水系滤膜过滤)。
(5)溶解度的测定。至少重复两次测定化合物在pbs缓冲液或水中的吸收峰面积,计算平均值,并代入到标准曲线方程中,计算相应的饱和溶解度。每测定完一个浓度,进样针应先用所用的色谱甲醇洗涤,再用下一个测试浓度溶液洗涤。
如图1所示,分别进样浓度为200μg/ml、40μg/ml、8μg/ml、1.6μg/ml和0.32μg/ml的gls4和i-5的标曲溶液,测量并计算得到gls4的标准曲线方程为y=31630x+83562,目标化合物i-5的标准曲线方程为y=15838x-99018;所建立的标准曲线方程具有较好的线性,r2分别为0.9979和0.9958。
表2i-5和gls4的水溶性
如表2所示,i-5在ph为7.4的pbs缓冲液中具有一定的溶解度(15.13μg/ml),远远高于母体药物gls4在ph为7.4的pbs缓冲液中的溶解度(<0.32μg/ml);i-5在水中的溶解度较小(<0.32μg/ml);由于待测样品的溶解度过小以及杂质的放大效应,i-5在水中的溶解度,gls4在ph为2.0的pbs缓冲液和水中的溶解度未检测出来。
实施例7.化合物i-5的急性毒性实验
(1)小鼠预处理。在山东大学动物实验中心购置15只体重在24~26g的成年雄性昆明小鼠,分为给药组、对照组和空白组,每组5只小鼠,将所有小鼠饲养在温度为25±1℃,湿度为60±5%的饲养室里。
(2)给药溶液的配制。将i-5依次溶于dmso(5%)、生理盐水(20%)和peg400(75%)中,超声至样品完全溶解;另配制不含i-5的同比例混合溶剂,4℃保存待用。
(3)小鼠的给药及给药后的观察。小鼠给药前先禁食24h,临近给药时对每只小鼠称重,给药组每只灌胃400μl样品溶液,对照组灌胃同体积不含样品的混合溶剂,空白组不做灌胃处理;每隔24h再对小鼠进行称重,同时监测其个体行为(嗜睡、阵挛性惊厥、厌食、皱皮等有无异常)。
如图2所示,对小鼠灌胃后,给药组4号小鼠灌胃后24h内死亡,对照组3号小鼠体重在一周观察期内基本不变,其余小鼠均在正常饮食下保持正常的生长状态,个体行为无异常,体重稳步增长,体重变化趋势用折线图表示。
实验结果表明,给药组小鼠7天内平均增重8.45g,对照组除3号小鼠外平均增重8.55g,给药组与对照组小鼠体重变化相比无明显差异;按最大剂量灌胃时i-5表现出较弱的急性毒性,ld50>2g/kg。
实施例8.化合物i-5的药代动力学实验
(1)样本及大鼠预处理。称量一定量的i-5于ep管中,加入计算量的dmso涡旋溶解,再加入计算量的peg400,涡旋,最后加入计算量的生理盐水。涡旋制剂使其混匀即得浓度为1.0mg/ml的给药制剂溶液。
10只健康的动物随机分成2组,所有动物禁食至少12h后称重,根据最新称量的动物体重计算给药量;给药后4h给予动物饲料。
(2)给药及采样。大鼠灌胃给药(iv:1mg/kg;po:10mg/kg)后,经颈静脉穿刺采血(采血点:给药前和给药后5min、15min、30min、1h、2h、4h、8h、10h、24h),每个样品采集约0.2ml全血/只/时间点,肝素钠抗凝,采集后放置冰上,1h之内离心分离血浆(离心条件:8000rpm,5min,8℃)。血浆样本在分析前存放于冷冻冰箱内。试验结束,所有存活动物按照医药科学委员会动物实验职业道德规定处死。
(3)样本的分析。样品采集结束后,采用lc-ms/ms法进行分析[column:thermoaccucorec18column(50×2.1mm,2.6μm);mobilephasea:h2o(0.1%formicacid);mobilephaseb:acn;flowrate:0.55ml/min];采用药代动力学软件phoenix8.0计算auc0-t、auc0-∞、mrt0-t和t1/2等参数。
表3hplc流动相条件
大鼠单次灌胃和静脉给药i-5后,不同时间点可以检测到血浆中gls4的浓度;而i-5本身在大鼠血浆中极不稳定,大鼠血浆中标准曲线无法成功,故其血药浓度无法获得。灌胃和静脉给药不同时间点血浆中gls4药代动力学参数个体值见表4,血药浓度-时间曲线见图3。
表4灌胃和静脉给药i-5后的主要药代动力学参数
由表4和图3可得,i-5在体内药代动力学性质较差,口服和静注给药后检测到的最高血药浓度分别为3.54ng/ml和7.48ng/ml,半衰期分别为0.5h和0.08h,静脉注射的生物利用度为10.11%,可能由于i-5在体内极不稳定,很快代谢为其他副产物,无法释放足够浓度的活性原药。
- 还没有人留言评论。精彩留言会获得点赞!