疼痛治疗中的CYP2J2拮抗剂的制作方法
本发明涉及对神经病理性疼痛、尤其是化疗诱导的外周神经病理性疼痛(CIPNP)的新疗法。本发明提供了细胞色素P450表氧化酶(CYP)的拮抗剂,更具体地,CYP2J2的拮抗剂,作为治疗剂用在诸如CIPNP的神经病理性疼痛的治疗中。CYP2J2拮抗剂已在体内鉴定为缓解CIPNP,并由此与化疗药组合而额外提供,用于治疗诸如癌症或其它增生性病症的疾病中。该CYP2J2拮抗剂减轻化疗诱导的疼痛,于是在癌症治疗期间允许使用更高和更佳的化疗剂量。此外,本发明涉及使用CYP2J2激动剂、或CYP2J2的代谢物,用于敏化TRPV1。这种情况下,本发明提议使用CYP2J2激动剂或代谢物与瞬时受体电位香草酸亚型1(TRPV1)激动剂的组合来治疗对TRPV1激动剂有应答的病症,例如神经病理性疼痛。
描述
神经病理性疼痛为持续的或慢性疼痛综合征,其可源自对神经系统、外周神经、背根神经节、背根、或者对中枢神经系统的损伤。神经病理性疼痛综合征包括异常性疼痛、诸如带状疱疹后神经痛和三叉神经痛的各种神经痛、幻觉痛、以及复杂性区域疼痛综合征,例如反射交感性营养不良和灼痛。灼痛通常的特征是,伴有痛觉过敏和异常性疼痛的自发烧灼痛。不幸的是,没有用于充分地、可预见地和特异性地治疗已出现的神经病理性疼痛的现有方法,由于用于神经病理性疼痛的目前治疗方法仅由试图帮助患者应对心理或作业疗法组成,而不是通过减轻或消除所承受的疼痛。对于神经病理性或者慢性疼痛的治疗对于医师和患者是一个挑战,因为没有药物特异地靶向该状况,以及因为目前使用的药物仅带来微小缓解并且基于它们在急性疼痛状况中的功效或者它们缓解副作用如焦虑和抑郁的功效。慢性疼痛的发病率在社会中是逐渐上升的,其对社会的负担在健康护理和生产力损伤方面都是巨大的。当前,没有经科学验证的缓解慢性疼痛的疗法。结果,卫生界致力于“疼痛管理”,其中同时使用多个模式疗法,希望对生活质量提供一些改善。因此,亟需可缓解慢性疼痛的药物。
化疗诱导的外周神经病理性疼痛(CIPNP)为严重的细胞抑制剂(例如紫杉烷类、铂衍生物、长春花生物碱类等)剂量限制性副作用。症状通常从麻刺感开始,并且可引起烧灼痛、刺痛和酸痛以及冷触发痛和机械性触痛。由于CIPNP,一些患者过早地停止了细胞抑制剂抗癌疗法,产生了更高的肿瘤进展风险。遗憾的是,很多已批准用于治疗不同的神经病理性疼痛的有前途的物质(例如加巴喷丁或阿米替林)在CIPNP的单一疗法中看起来很少或者没有镇痛作用。对细胞和分子机制的了解对于治疗或甚至预防CIPNP是必要的,可以改进细胞抑制剂疗法的总成功率。
最近的研究鉴定出离子通道的瞬时受体电位家族的成员(TRPV1、TRPA1和TRPV4)在奥沙利铂和紫杉醇诱导的神经病理期间作为机械性触痛和冷触发痛的促进者。激活或敏化TRPV1和TRPA1可导致CGRP和物质P的增强释放,这二者都可引起神经源性炎症和T细胞的募集。
但是,仍不清楚哪种内源性介质参与了细胞抑制剂依赖的TRP通道激活或敏化,由于没有细胞抑制剂可直接激活TRP通道。有趣的是,紫杉醇和奥沙利铂都为CYP表氧化酶的诱导剂(紫杉醇:CYP2C8、CYP2C9,奥沙利铂:CYP2E1、CYP1B1)。细胞色素P450(CYP)-表氧化酶可代谢ω-6脂肪酸,例如花生四烯酸(AA)和亚油酸(LA),产生脂环氧化物如EETs(环氧二十碳三烯酸)或ω-羟化物如20-HETE。
细胞色素P450单加氧酶花生四烯酸的代谢导致各种生物活性的类花生酸的形成。已知发生三种类型的氧化反应。第一,烯烃环氧化反应(表氧化酶催化)产生环氧二十碳三烯酸(EETs)。四种重要的EET区域异构体为[5,6]-EET、[8,9]-EET、[11,12]-EET、和[14,15]-EET。这些EET经环氧化物水解酶水解形成对应的二羟基二十碳三烯酸(DHET)。第二,ω末端氧化导致形成ω末端羟基二十碳三烯酸(HETE)。第三,烯丙位氧化导致形成中链HETE。
已经鉴定了几种细胞色素P450表氧化酶,包括CYP1A、CYP2B、CYP2C、CYP2E和CYP2J亚族的成员。人们最近关注CYP2J亚族的蛋白。CYP2J的一个具体亚型CYP2J2在人心肌细胞中高度表达,在这里花生四烯酸经代谢产生EET。CYP2J2蛋白也在气管和肠道中的上皮细胞内被发现。与其它P450酶比较,CYP2J2蛋白沿肠道径向在上皮细胞和非上皮细胞中均匀分布。在自主神经节的细胞中、上皮细胞中、以及肠平滑肌细胞中发现高水平的CYP2J2蛋白。在动物中已鉴定了几种CYP2J同源物,包括大鼠CYP2J3、大鼠CYP2J4、小鼠CYP2J5和小鼠CYP2J6。
辣椒素为瞬时受体电位香草酸亚型1受体(TRPV1;之前也称为香草酸受体1(VR1))的高度选择性激动剂,该受体为配体门控的非选择性阳离子通道,优先在较小直径的感觉神经元上表达,特别是那些专门检测疼痛或有害感觉的C纤维上。TRPV1应答有害刺激,包括辣椒素、热、以及细胞外酸化,并且将整合同时暴露于这些刺激。对表达TRPV1的(辣椒素敏感的)伤害感受器的活化的最初效果为灼烧感觉、痛觉过敏、异常性疼痛、以及红斑。但是,在持续暴露于低浓度辣椒素或单次暴露于高浓度辣椒素或其它TRPV1激动剂后,小直径的感觉轴突变得对各种刺激(包括辣椒素或热刺激)较不敏感。这种持续暴露也以减弱的疼痛应答为特征。辣椒素的这些后期阶段效应经常被称为“脱敏”,为开发局部辣椒素制剂来治疗各种疼痛综合征和其它情况的基本原理。
因此,辣椒素、辣椒素类物质和TRPV1激动剂可以用于缓解多种疾病。例如,它们可以用于治疗神经病理性疼痛(包括与糖尿病性神经病变、带状疱疹后神经痛、HIV/AIDS、创伤性损伤、复杂性区域疼痛综合征、三叉神经痛、红斑性肢痛和幻觉痛相关的疼痛)、混合伤害性和/或神经病理性混合病因产生的疼痛(例如,癌症)、骨关节炎、纤维肌痛、腰痛、炎性痛觉过敏、外阴前庭炎或外阴疼痛、窦息肉间质性膀胱炎、神经源性膀胱或膀胱过动症、前列腺增生、鼻炎、手术、创伤、直肠过敏、灼口综合症、口腔黏膜炎、疱疹(或其它病毒感染)、前列腺肥大、皮炎、瘙痒、痒、耳鸣、牛皮癣、疣、癌症(特别是皮肤癌)、头疼、以及皱纹。
因此,迄今为止,没有可用于神经病理性疼痛的特异疗法,特别是针对化疗诱导的外周神经病理性疼痛(CIPNP),这限制了癌症治疗期间化疗剂的最大剂量,并且给经历化疗的患者的生活质量的带来严重损害。因此本发明的目的为提供新的治疗选择来应对神经病理性疼痛,特别是CIPNP。
在第一方面,通过将细胞色素P450表氧化酶(CYP)拮抗剂用于在受试者中预防或治疗疼痛来解决上述问题。在本发明的一些实施方案中,该CYP拮抗剂选自CYP1A、CYP2B、CYP2C、CYP2E、以及优选的CYP2J拮抗剂。最优选地,该CYP拮抗剂为CYP2J2的哺乳动物同源物的拮抗剂(CYP2J2拮抗剂),优选人CYP2J2的拮抗剂,例如替米沙坦、阿立哌唑或最优选特非那定。
本发明涵盖任何CYP2J2拮抗剂(优选选择性CYP2J2拮抗剂)的用途。术语“选择性CYP2J2拮抗剂”涉及选择性抑制CYP2J2而非其它相关酶如CYP3A分子的活性、功能或表达的CYP2J2拮抗剂。为了鉴定候选拮抗剂是否为CYP2J2拮抗剂,可采用发光的细胞色素P450发光测定法。CYP蛋白催化花生四烯酸代谢物的形成。发光的CYP测定使用用于荧光素酶的生光反应的底物前体。CYP将该底物前体转化为荧光素或者荧光素酯,其在与称为荧光素检测试剂(LDR)的荧光素酶反应混合物的第二反应中生成光。在该第二反应中产生的光的量与CYP活性成比例。
为了测试候选CY2J2拮抗剂的选择性,可采用特异于其它CYP酶(如CYP3A4)的发光CYP测定。将候选拮抗剂针对CYP2J2与该相同拮抗剂针对另一CYP蛋白(如CYP3A4)的抑制活性进行比较,由此提供关于候选拮抗剂的选择性的信息。
在本发明上下文中,优选的选择性CYP2J2拮抗剂选自本文新公开的CYP2J2拮抗剂:雌二醇、盐酸酚苄明、氯雷他定、丙酸氯倍他索、多沙唑嗪甲磺酸盐、非诺贝特、左炔诺孕酮、阿立哌唑、哈西奈德、替米沙坦、氯苯吩嗪、左旋甲状腺素钠、阿洛司琼盐酸盐、醋酸氟轻松、碘塞罗宁钠、美克洛嗪二盐酸盐和特非那定以及它们的衍生物。
在本文描述的发明的背景下,所述待治疗的疼痛优选神经病理性疼痛(包括与糖尿病性神经病变相关的疼痛、带状疱疹后神经痛、HIV/AIDS诱导的神经病理性疼痛、创伤性损伤、复杂性区域疼痛综合征、三叉神经痛、红斑性肢痛和幻觉痛)、混合伤害性和/或神经病理性混合病因产生的疼痛(例如,癌症)、骨关节炎、纤维肌痛、腰痛、炎性痛觉过敏、外阴前庭炎或外阴疼痛、窦息肉间质性膀胱炎、神经源性膀胱或膀胱过动症、前列腺增生、鼻炎、手术、创伤、直肠过敏、灼口综合症、口腔黏膜炎、疱疹(或其它病毒感染)、前列腺肥大、皮炎、瘙痒、痒、耳鸣、牛皮癣、疣、癌症、头疼、以及皱纹、由于中风或块状病变、脊髓损伤、或多发性硬化导致的中枢疼痛。但是,最优选的实施方案涉及化疗诱导的外周神经病理性疼痛(CIPNP)。
本发明在此提供了疼痛疗法,其包括抑制尤其是CYP2J2的活性,而CYP2J2产生代谢化合物9,10-EpOME—根据本发明,该化合物为离子通道介导的疼痛感觉的敏化剂。令人惊讶地,根据本发明,对CYP2J2的抑制证实在体内是有效的,缓解了在小鼠模型中通过紫杉醇诱导的神经病理性疼痛,表明了CYP2J2拮抗剂作为针对神经病理性疼痛尤其是CIPNP的止痛剂的用途。
本发明的一个另外实施方案涉及以上提到的疼痛的预防或治疗,其包括给患有所述疼痛的受试者施用本发明的所述CYP拮抗剂,其中所述受试者接受了、正接受或将接受化疗。因此,该受试者在优选的实施方案中为患有或诊断为具有癌症疾病的受试者。
在本发明上下文中,化疗优选包括向有需要治疗的受试者施用化疗剂,该化疗剂选自基于嘧啶酮的抗肿瘤药,例如阿糖胞苷,5-氟尿嘧啶或铂类药物,例如顺铂,或紫杉烷类,例如紫杉醇、多西他赛或卡巴他赛,或者它们的衍生物。这些化疗剂已知诱导神经病理性疼痛,尤其对于紫杉烷类是已知的,因此它们在本发明上下文中是优选的。最优选的是紫杉醇。
此外,根据本发明,所述疼痛的预防或治疗包括同时或顺序施用所述CYP拮抗剂和所述化疗剂。对于这个实施方案,还参见以下对本发明的组合的描述。
本发明的问题在另一方面是通过用于预防或治疗受试者中的疼痛的9,10-环氧-12Z-十八碳烯酸(9,10-EpOME)拮抗剂来解决的。发现9.10-EpOME是通过CYP活性而产生的。因此,代替拮抗CYP,本发明的结果可以通过直接拮抗9,10-EpOME以避免敏化介导疼痛的神经元而以另外方式实现。本发明的这些9,10-EpOME拮抗剂优选为小分子(但也可为蛋白或肽(例如,抗体或其片段)),其结合9,10-EpOME并抑制对TRPV1的敏化。
就此而言,以上描述的CYP拮抗剂的用途的具体实施方案也适用于本发明的9,10-EpOME拮抗剂,特别是涉及所述预防或治疗以及化疗的实施方案。
该问题又通过一组合来实现,该组合包括(i)CYP拮抗剂或9,10-EpOME拮抗剂以及(ii)用于在预防或治疗疾病中同时或顺序使用的化疗剂,其中该疾病选自增生性病症,例如癌症,或疼痛,例如CIPNP。
在本文中,术语“增生性病症”在广义上包括任何需要控制细胞周期的病症,例如心血管病症如再狭窄和心肌病,自身免疫病症如肾小球肾炎和类风湿性关节炎,皮肤病症如牛皮癣,抗炎、抗真菌、抗寄生物病症如疟疾、肺气肿和脱发。在这些病症中,本发明的化合物可以根据需要在期望的细胞中诱导凋亡或保持停滞。优选地,该增生性病症为癌症或白血病,最优选乳腺癌、肺癌、前列腺癌、膀胱癌、头颈癌、结肠癌、卵巢癌、子宫癌、肉瘤或淋巴瘤。
本实施方案还涉及治疗患有疼痛的受试者组,其中受试者在接受化疗剂治疗。因此,本发明的CYP拮抗剂可以在癌症治疗的同时期施用,或者在之前或之后进行,这可为优选的,以避免累积的不良反应。当本发明的CYP拮抗剂的生理效应和化疗剂的疼痛诱导在有需要这种治疗的受试者中组合时,实现了本发明的结果。在治疗期间的最后一剂药物给药后,该药物诱导的生理效应将不会立即消失,而最可能是之后消失。因此,在顺序的治疗周期中使用本发明的拮抗剂,例如本发明的拮抗剂不是同时而是在化疗之前给药,则仍在患者内导致两种化合物的临床效果的组合,因此落入本发明的组合治疗的含义内。
术语“组合”在本上下文中指制剂中的两种或更多种活性物质的活性物质组合,也指治疗性处理中彼此以特定间隔施用的活性物质各个制剂意义上的组合。因此,术语“组合”应包括两种或多种治疗上有效的化合物的共给药,如在本发明上下文中所描述的。
共施用:在本申请上下文中,两种或更多种化合物的共施用定义为在一年内给患者施用两种或更多种化合物,包括分开施用两种或更多种药物(每种含有化合物的一种),以及同时施用,无论两种或更多个化合物是组合在一种制剂中,还是它们处于两种或更多种分开的制剂中。
在一个实施方案中,本发明的组合包括(i)和(ii),通过在所述预防或治疗期间给受试者顺序或同时施用来组合,优选其中拮抗剂和化疗剂在所述预防或治疗期间同时施用。
化疗剂优选选自基于嘧啶酮的抗肿瘤药,例如阿糖胞苷、5-氟尿嘧啶,或铂剂,例如顺铂,或紫杉烷类,例如紫杉醇或多西他赛,或它们的衍生物。最优选的化疗剂为紫杉醇或多西他赛。
本文描述的发明的拮抗剂优选选自由抑制性RNA、抑制性抗体或其片段、和/或小分子组成的化合物组。下文提供了对优选的CYP拮抗剂的详细描述。
在本发明上下文中,还优选给所述受试者施用至少一种另外的对疼痛有效的治疗剂,例如吗啡、阿片类或非阿片类镇痛剂或其它镇痛剂。
在本发明的另一方面,提供了在受试者中预防或治疗疼痛的方法,该方法包括给所述受试者施用治疗有效量的本发明的CYP拮抗剂或9,10-EpOME拮抗剂的步骤。该CYP拮抗剂优选选自CYP1A、CYP2B、CYP2C、CYP2E和CYP2J拮抗剂。该CYP2J拮抗剂优选为CYP2J2的哺乳动物同源物的拮抗剂(CYP2J2拮抗剂),优选人CYP2J2的拮抗剂,例如特非那定、替米沙坦、以及它们的生物类似物或衍生物。
其它优选的CYP拮抗剂选自雌二醇、盐酸酚苄明、氯雷他定、丙酸氯倍他索、多沙唑嗪甲磺酸盐、非诺贝特、左炔诺孕酮、阿立哌唑、哈西奈德、替米沙坦、氯苯吩嗪、左旋甲状腺素钠、阿洛司琼盐酸盐、醋酸氟轻松、碘塞罗宁钠、美克洛嗪二盐酸盐和特非那定。
在前述方法背景下可治疗的疾病在上文中有描述。
在治疗或预防期间,优选给所述患者施用至少一种另外的对疼痛有效的治疗剂,例如其它的镇痛剂,例如阿片类或非阿片类镇痛剂。
接着,本发明的另外方面涉及增加受试者中瞬时受体电位香草酸亚型1(TRPV1)的敏感度的方法,包括给所述受试者施用治疗有效量的9,10-EpOME或CYP2J2激动剂。
在本发明背景下,令人惊讶地发现9,10-EpOME敏化TRPV1通道蛋白,而其为疼痛感觉的主要介质。因此,在优选的实施方中,本发明提供了作为TRPV1激动剂的9,10-EpOME,这在医药中是尤其有用的。9,10-EpOME和TRPV1激动剂辣椒素的组合显著增强辣椒素活性。一个实施方案例如涉及治疗以病理性抑制的疼痛感觉或以疼痛不敏感为特征的疾病。
在另一实施方案中,9,10-EpOME可以用在增强TRPV1激动剂如辣椒素的活性的方法中。辣椒素作为镇痛剂用在局部软膏、鼻喷剂、以及皮肤贴剂中以缓解疼痛。其可以以乳膏形式应用,用于暂时缓解肌肉和关节炎受累关节的小痛和疼痛、背痛、拉伤和扭伤,通常在具有其他发红剂的化合物中。其还用于减轻外周神经病变的症状,例如由带状疱疹引起的带状疱疹后神神经痛。
辣椒素的镇痛和/或抗炎效果的发生机制通过模拟烧灼感觉而得到支持;通过钙内流压倒神经,导致伤害感受器脱敏和/或凋亡,并由此使得神经在增长的时间段内不能报告疼痛。长期暴露于辣椒素下,神经元的伤害感受器经历凋亡,导致疼痛感觉减弱并阻断神经源性炎症。如果去除了辣椒素,则伤害感受性神经元随时间逐渐恢复。因此,本发明的9,10-EpOME的使用可以大大增强辣椒素和相关化合物的医疗效果,或者可以帮助减少辣椒素剂量。
因此,在本发明的优选实施方案中,提供了在受试者中治疗疾病的方法,包括施用治疗有效量的(i)9,10-EpOME或CYP2J2激动剂和(ii)TRPV1激动剂。至于顺序或同时使用治疗剂,可参照上文的说明,它们一样地应用于本发明的这个方面。
疾病优选选自神经病理性疼痛(包括与糖尿病性神经病变、带状疱疹后神经痛、HIV/AIDS、创伤性损伤、复杂性区域疼痛综合征、三叉神经痛、红斑性肢痛和幻觉痛相关的疼痛)、混合伤害性和/或神经病理性混合病因产生的疼痛(例如,癌症)、骨关节炎、纤维肌痛、腰痛、炎性痛觉过敏、外阴前庭炎或外阴疼痛、窦息肉间质性膀胱炎、神经源性膀胱或膀胱过动症、前列腺增生、鼻炎、手术、创伤、直肠过敏、灼口综合症、口腔黏膜炎、疱疹(或其它病毒感染)、前列腺肥大、皮炎、瘙痒、痒、耳鸣、牛皮癣、疣、癌症、头疼、以及皱纹。通常,包括任何可通过TRPV1激动剂治疗的疾病。
本发明的示例性和优选的TRPV1激动剂选自辣椒素、胡椒碱、6-姜酚、6-姜烯酚、α-山椒素、β-山椒素、γ-山椒素、δ-山椒素、羟基α-山椒素以及羟基β-山椒素。
于是,另一方面涉及9,10-EpOME或CYP2J2激动剂在上述方法中的应用。
再一方面涉及(i)9,10-EpOME或CYP2J2激动剂和(ii)TRPV1激动剂的组合在医药中的应用,优选在治疗选自神经病理性疼痛(包括与糖尿病性神经病变、带状疱疹后神经痛、HIV/AIDS、创伤性损伤、复杂性区域疼痛综合征、三叉神经痛、红斑性肢痛和幻觉痛相关的疼痛)、混合伤害性和/或神经病理性混合病因产生的疼痛(例如,癌症)、骨关节炎、纤维肌痛、腰痛、炎性痛觉过敏、外阴前庭炎或外阴疼痛、窦息肉间质性膀胱炎、神经源性膀胱或膀胱过动症、前列腺增生、鼻炎、手术、创伤、直肠过敏、灼口综合症、口腔黏膜炎、疱疹(或其它病毒感染)、前列腺肥大、皮炎、瘙痒、痒、耳鸣、牛皮癣、疣、癌症(特别是皮肤癌)、头疼、以及皱纹的疾病中的应用。
该TRPV1激动剂优选选自辣椒素、胡椒碱、6-姜酚、6-姜烯酚、α-山椒素、β-山椒素、γ-山椒素、δ-山椒素、羟基α-山椒素以及羟基β-山椒素。
根据本文描述的发明,受试者优选哺乳动物,优选人,最优选接受化疗治疗的人,例如癌症患者。
CYP-拮抗剂
在本发明上下文中,“CYP拮抗剂”优选选自CYP1A、CYP2B、CYP2C、CYP2E、以及更优选的CYP2J拮抗剂。最优选地,该CYP拮抗剂为CYP2J2的哺乳动物同源物的拮抗剂(CYP2J2拮抗剂),优选人CYP2J2的拮抗剂。因此,在本文描述的发明的最优选的实施方案以及方面中,术语“CYP拮抗剂”为CYP2J2拮抗剂或人CYP2J2的哺乳动物同源物的拮抗剂。
在本文中,术语“CYP拮抗剂”指引起CYP表达或活性的量或速率降低的物质。这种物质可直接例如通过结合CYP和降低CYP表达或活性的量或速率来起作用。CYP拮抗剂还可降低CYP表达或活性的量或速率,例如通过结合CYP以使得减弱或防止CYP与CYP受体的相互作用;通过结合并修饰CYP,例如通过去除或添加一部分;以及通过结合CYP并降低其稳定性。CYP拮抗剂也可间接地起作用,例如通过结合调节分子或基因区域以调控调节蛋白或基因区域功能,并引起CYP表达或活性的量或速率的降低。因此,CYP拮抗剂可通过任何导致CYP表达或活性的量或速率的降低的机制起作用。
CYP拮抗剂可例如为天然或非天然存在的大分子,例如多肽、肽、肽模拟物、核酸、碳水化合物或脂质。CYP拮抗剂还可为抗体、或其抗原结合片段,例如单克隆抗体、人源化或人抗体、嵌合抗体、微抗体、双功能抗体、单链抗体(scFv)、可变区片段(Fv或Fd)、Fab或F(ab)2。CYP拮抗剂还可为特异于CYP的多克隆抗体。CYP拮抗剂还可为天然存在的大分子的部分或完全合成的衍生物、类似物或模拟物,或小有机或无机分子。
作为抗体的CYP拮抗剂可例如为结合CYP并抑制与CYP受体结合的抗体,或者改变调节CYP表达或活性的分子的活性以使CYP表达或活性的量或速率降低的抗体。用在本发明方法中的抗体可为天然存在的抗体,包括单克隆或多克隆抗体或其片段,或者非天然存在的抗体,包括但不限于,单链抗体、嵌合抗体、双功能抗体、互补决定区移植(CDR移植)抗体和人源化抗体或它们的抗原结合片段。
作为核酸的CYP拮抗剂可例如为反义核苷酸序列、RNA分子、或适体序列。反义核酸序列可结合细胞内的核苷酸序列并调控CYP的表达水平,或者调控另一控制CYP的表达或活性的基因的表达。类似地,诸如催化性核酶的RNA分子可结合并改变CYP基因或控制CYP的表达或活性的其它基因的表达。适体为具有能够结合分子靶标的三维结构的核酸序列。
作为核酸的CYP-拮抗剂还可为用在RNA干扰方法中的双链RNA分子。RNA干扰(RNAi)为通过转录后RNA降解进行的序列特异性基因沉默过程,其由在序列上与所沉默的基因同源的双链RNA(dsRNA)起始。适用于RNAi的双链RNA含有对应待靶向基因的约21个连续氨基酸的正义和反义链,它们形成19个RNA碱基对,在每个3'末端留下两个核苷酸的突出端(Elbashir等,Nature 411:494-498(2001);Bass,Nature 411:428-429(2001);Zamore,Nat.Struct.Biol.8:746-750(2001))。约25至30个核苷酸的dsRNA也已成功的用于RNAi(Karabinos等,Proc.Natl.Acad.Sci.USA 98:7863-7868(2001)。dsRNA可在体外合成,并通过本领域的已知方法导入细胞。
优选的CYP2J2拮抗剂选自雌二醇、盐酸酚苄明、氯雷他定、丙酸氯倍他索、多沙唑嗪甲磺酸盐、非诺贝特、左炔诺孕酮、阿立哌唑、哈西奈德、替米沙坦、氯苯吩嗪、左旋甲状腺素钠、阿洛司琼盐酸盐、醋酸氟轻松、碘塞罗宁钠、美克洛嗪二盐酸盐和特非那定。
治疗或预防疼痛或其它神经病症的组合物和试剂盒。
本申请的另一方面涉及通过采用本发明的化合物或组合来治疗或预防疼痛或增生性病症的组合物和试剂盒。在一个实施方案中,该组合物包括上文描述的化合物,其中所述化合物优选选自抗体、抗体片段、短干扰RNA(siRNA)、适体、合成抗体(synbody)、结合剂、肽、适体-siRNA嵌合体、单链反义寡核苷酸、三链体形成寡核苷酸、核酶、外在引导序列、试剂编码表达载体(agent-encoding expression vector)、小分子以及药学上可接受的载体。
在本文中,用语“药学上可接受的载体”旨在包括与药物施用相符的任何和所有的溶剂、增溶剂、填充剂、稳定剂、粘结剂、吸附剂、碱、缓冲剂、润滑剂、控释载体、稀释剂、乳化剂、湿润剂、润滑剂、分散介质、涂层、抗菌或抗真菌剂、等张和吸收延缓剂,等等。将这些介质和试剂用于药学上有活性的物质在本领域内是公知的。除了任何常规媒介物或试剂与该活性化合物不相容之外,考虑将其用在该组合物中。补充剂也可掺入到该组合物中。在某些实施方案中,该药学上可接受的载体包括血清白蛋白。
本发明的药物组合物配制为与其预期的施用途径相容。施用途径的实例包括胃肠外施用,例如鞘内、动脉内、静脉内、皮内、皮下施用,口服,透皮(局部)和跨粘膜施用。
用于胃肠外、皮内或皮下应用的溶液或混悬液可包括以下组分:无菌稀释剂如注射用水,盐水溶液,不挥发油,聚乙二醇,甘油,丙二醇或其它合成溶剂;抗菌剂例如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂例如抗坏血酸或硫酸氢钠;螯合剂例如乙二胺四乙酸;缓冲剂例如乙酸盐,柠檬酸盐或磷酸盐以及用于调节张力的试剂如氯化钠或葡萄糖。可用酸或碱调节pH,例如盐酸或氢氧化钠。胃肠外配制物可封在安瓿瓶、一次性注射器或多重剂量小瓶中,它们由玻璃或塑料制成。
适合于可注射使用的药物组合物包括无菌水溶液(当水溶时)或混悬液以及用于临时制备无菌可注射溶液或混悬液的无菌粉末。对于静脉内施用,适合的载体包括生理盐水、抑菌水、商品名为Cremophor ELTM的物质(BASF,Parsippany,N.J.)或磷酸盐缓冲盐水(PBS)。所有情况下,可注射组合物应为无菌的,并且应为流体,其程度为易于注射。其在生产和保存条件下必须是稳定的,并且必须对微生物如细菌和真菌的污染作用防腐。载体可为溶剂或分散介质,含有例如水、乙醇、多元醇(例如,甘油、丙二醇、以及液体聚乙二醇,等)、或它们的适当混合物。例如通过使用诸如卵磷脂的涂层、对于混悬液的情况通过维持所需的粒径以及通过使用表面活性剂可维持适当的流动性。预防微生物作用可通过各种抗菌和抗真菌剂来实现,例如,对羟基苯甲酸酯、氯丁醇、酚、抗坏血酸、硫柳汞,等。很多情况下,优选在组合物中包括等张剂,例如,蔗糖、多元醇如甘露醇、山梨醇和氯化钠。可注射组合物的长时间吸收可通过在组合物中包括进延缓吸收的试剂来实现,例如单硬脂酸铝和明胶。
可通过将活性化合物(例如,神经调节蛋白)以所需量与以上列出的成分的一种或组合掺入适合的溶剂中,然后无菌过滤(需要的话),来制备无菌可注射溶液。通常,通过将活性化合物掺入含有基础分散介质和所需的来自以上列出成分的其它成分的无菌载体中来制备分散体。对于用于制备无菌可注射溶液的无菌粉末的情况,优选的制备方法为真空干燥和冷冻干燥,其产生活性成分加上任何额外所需成分(来自其之前无菌过滤的溶液)的粉末。
口服组合物通常包括惰性稀释剂或可食用载体。它们可包封在明胶胶囊中或压缩为片剂。出于口服治疗施用的目的,活性化合物可与赋形剂一起掺混,并以片剂、含片、或胶囊的形式使用。口服组合物还可采用流体载体来制备,以用作漱口水,其中流体载体中的化合物口服施用,在口中来回,并且吐出或咽下。药学上相容的粘合剂和/或佐剂材料可作为组合的一部分包括进来。片剂、小丸、胶囊、含片等可含有以下成分或类似性质化合物的任何一种:粘合剂如微晶纤维素、黄蓍胶或明胶;赋形剂如淀粉或乳糖;崩解剂如褐藻酸、羟基乙酸淀粉钠(Primogel)、或玉米淀粉;润滑剂如硬脂酸镁或斯泰特(Stertes);助流剂如胶体二氧化硅;甜味剂如蔗糖或糖精;或调味剂如薄荷、水杨酸甲酯、或橙香精。
对于通过吸入施用,化合物是以气溶胶喷雾的形式从含有适合的推进剂(例如,诸如二氧化碳的气体)的压力容器或分配器或喷雾器递送。
全身施用也可通过跨粘膜或透皮手段进行。对于跨粘膜或透皮施用,将适合于待渗透屏障的渗透剂用在制剂中。这些渗透剂通常在本领域是已知的,包括例如用于跨粘膜施用的洗涤剂、胆汁盐、以及梭链孢酸衍生物。跨粘膜施用可通过使用鼻喷剂或者栓剂来完成。对于透皮施用,药物组合物配制为软膏、药膏、凝胶、或乳膏,如本领域所公知的。
在某些实施方案中,药物组合物配制为活性成分的持续或控制释放。可使用生物可降解的、生物相容的聚合物,例如乙烯醋酸乙烯酯、聚酸酐、聚乙醇酸、胶原、聚原酸酯、以及聚乳酸。用于制备这些制剂的方法对本领域技术人员是显而易见的。这些材料也可在商业上从例如从阿尔扎(Alza Corporation)和诺华制药公司(Nova Pharmaceuticals,Inc.)获得。脂质体混悬液(包括带有针对病毒抗原的单克隆抗体的靶向感染细胞的脂质体)也可用作药学上可接受的载体。这些可按照本领域技术人员已知的方法来制备。
特别有利的是,配制单位剂量形式的口服或胃肠外组合物,以方便施用和剂量一致性。单位剂量形式用在本文时包括物理上分开的单位,适合作为单一剂量用于待治疗的受试者;每个单位含有预定量的活性化合物,经计算与所需的药物载体联合会产生期望的治疗效果。本发明单位剂量形式的规格取决于并直接依赖于活性化合物的独特特征和待获得的具体治疗效果,以及本领域中配制这种用于个体治疗的活性化合物的内在限制。
这些化合物的毒性和治疗功效可通过标准药学程序在细胞培养或试验动物中确定,例如用于确定LD50(50%群体致死剂量)和ED50(50%群体治疗上有效的剂量)。毒性和治疗效果的剂量比例为治疗指数,其可表示为比例LD50/ED50。显示出大治疗指数的化合物是优选的。尽管可以使用显示出毒性副作用的化合物,但应考虑设计将这些化合物靶向受累组织位点的递送系统,以便对非感染细胞的潜在损伤降至最小,并由此减小副作用。
从细胞培养测定和动物研究获得的数据可用在制定用于人的剂量范围中。这些化合物的剂量优选处于包括具有微小毒性或没有毒性的ED50的循环浓度的范围内。剂量可以在该范围内随采用的剂型和使用的施用途径而变化。对于用在本发明方法中的任何化合物,治疗有效剂量可从细胞培养测定初步估计。可在动物模型中制定获得包括在细胞培养中确定的IC50(即,实现症状的半数最大抑制的测试化合物的浓度)的循环血浆浓度范围的剂量。这种信息可用于更精确地确定人类可用剂量。药物组合物可连同施用说明书包括在容器、包装盒、或分散器中。
现在将参照附图和序列在以下实施例中进一步描述本发明,然而并不限于此。出于本发明的目的,本文提到的所有参考文献都通过引用将它们的全文并入。在附图中:
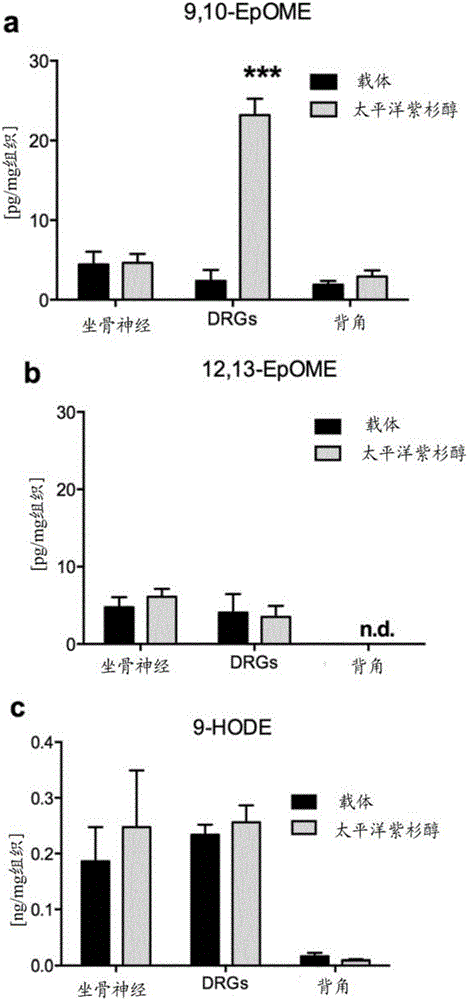
图1:紫杉醇CIPNP或炎症期间氧化的亚油酸代谢物的浓度。所显示的为C57B16/N小鼠i.p.注射载体(黑色)或紫杉醇(灰色,6mg·kg-1)24h后,坐骨神经、DRG和脊髓背角中9,10-EpOME(a)和12,13-EpOME(b)的浓度;n.d.:未检测到。C57B16/N小鼠i.p.注射载体(黑色)或紫杉醇(灰色)24h后,坐骨神经、L4-L6-DRGs和对应的脊髓背角节段中9-HODE(c)和13-HODE(d)的浓度。(e)足底内注射酵母聚糖(12.5mg/ml,20μl)24h后,L4-L6-DRGs和背角的相应节段中的9,10-EpOME浓度关系。数据表示每组8至10只动物的均值±SEM;***p<0.001,学生t检验。
图2:9,10-EpOME对DRG神经元的直接作用。9,10-EpOME[10μM,30s]的应用在DRG神经元上引起钙瞬变,其应答于高钾(50mM KCl,30s)。显示了代表性描记线。(b)DRG神经元中9,10-EpOME依赖的钙相对于应答神经元数量增加的剂量反应关系。数据表示每个浓度5次测量的均值±SEM。(c)和(d)在9,10-EpOME刺激前和后2分钟采用含有EGTA(2mM)的无钙培养基洗入,可阻断9,10-EpOME[10μM,30s]引起的钙瞬变。数据表示24个(无钙)或16个(对照)神经元的均值±SEM。(e)和(f)9,10-EpOME[10μM,30s]的钙瞬变可被选择性TRPV1拮抗剂(AMG 9810,1μM)而不是选择性TRPA1拮抗剂(HC-030031,20μM)阻断,这些阻断剂在第二次9,10-EpOME刺激前2分钟洗入。数据表示16个(对照)、31个(AMG 9810)或18个(HC-030031)神经元的均值±SEM;**p<0.01,学生t检验。
图3:9,10-EpOME在DRG神经元中剂量依赖性敏化TRPV1,并在脊髓薄片的第二层神经元中增强辣椒素诱导的自发性EPSC频率的增加。(a)用辣椒素双刺激DRG神经元(200nM,每次15s),并在第二次辣椒素刺激前用载体或9,10-EpOME[1μM]温浴2分钟。(b)采用与(a)中描述相同的规程,第一次和第二次辣椒素应答之间比率的剂量依赖性差异。数据表示以下数量神经元的均值±SEM:27个(对照)、26个(250nM 9,10-EpOME)、21个(500nM 9,10-EpOME)、19个(750nM 9,10-EpOME)、41个(1μM 9,10-EpOME)、18个(2μM 9,10-EpOME)、或28个(采用50μM AITC 20s代替辣椒素);*p<0.05,**p<0.01,***p<0.001学生t检验。(c)自发性EPSC(sEPSC)在第二层神经元中的描记线。下方插图,1、2、3和4为放大的,分别表示对基线、第一辣椒素(1mM)、9,10-EpOME(1mM)、以及第二辣椒素(1mM)加9,10-EpOME的记录。(d)sEPSC的频率。与基线sEPSC相比,辣椒素诱导了sEPSC频率的深入增加(从6.9±0.4Hz和13.7±0.4Hz)。单独的9,10-EpOME治疗稍稍增加sEPSC频率(8.2±0.8Hz),并由辣椒素显著增强了该sEPSC频率增加(18.7±1.1Hz)。*P<0.05,与无治疗基线相比;#P<0.05,与第一次辣椒素治疗(1mM)相比。n=5个神经元/组。(E)sEPSC的幅度。辣椒素和9,10-EpOME对sEPSC幅度没有显著影响。n=5个神经元/组。
图4:9,10-EpOME在DRG神经元中对TRPV1的敏化由Gs偶联受体和cAMP-PKA途径所介导。(a)9,10-EpOME在大鼠DRGs中膜碎片中催化[y-35S]-GTP结合。在30μM GDP和载体(乙酸甲酯,0.7%(v/v))、腺苷[10μM]或9,10-EpOME[1μM]存在30分钟下,采用大鼠DRGs的膜碎片进行实验。从来自总共15只动物的膜碎片的3次测量获得数据。对于每次测量,将来自5只动物的DRG合并;*p<0.05,**p<0.01,使用唐恩(Dunn’s)多重比较事后检验的克鲁斯卡-沃利斯(Kruskal-Wallis)检验。(b)在9,10-EpOME、西卡前列素或毛喉素(每种1μM)刺激15分钟后富含神经元的DRG培养物中的cAMP浓度。数据表示来自5只小鼠的DRG培养物的均值±SEM。(c)和(d)9,10-EpOME[1μM]对TRPV1的敏化可通过以PKA抑制剂(H89-二盐酸盐,10μM,1h)预温浴而减弱。数据表示15个(载体)、19个(EpOME)或33个神经元(EpOME和H89预温浴)的均值±SEM。(e)和(f)9,10-EpOME[1μM]对TRPV1的敏化不受PKC抑制剂预温浴(GF 109203X,10μM,1h)的影响。数据表示18个(载体)、23个(EpOME)或39个神经元(EpOME和GFX预温浴)的均值±SEM;*p<0.05,**p<0.01学生t检验;n.s.非显著的。
图5:足底内或鞘内注射9,10-EpOME减弱了野生型小鼠中的疼痛阈值并敏化辣椒素诱导的机械阈值。(a)和(b)C57B1/6N小鼠接受足底内注射9,10-EpOME(10μM)或载体(DMSO盐水溶液,0.3%(v/v))。在注射后5h监测热(a)或机械(b)阈值。数据表示来自8只小鼠的均值±SEM。(c)和(d)野生型BL/6N小鼠鞘内注射9,10-EpOME(10μM)或载体(DMSO盐水溶液,0.3%(v/v))。在注射后监测热(c)或机械(d)阈值2h(热)或3h(机械),第一小时间隔15分钟,第二小时间隔30分钟。数据表示来自8只小鼠的均值±SEM。图中未包括。
图6:分离的坐骨神经或富含神经元的DRG培养物在9,10-EpOME刺激后释放iCGRP。(a)从野生型BL/6N小鼠的分离的坐骨神经释放iCGRP,用以下溶液刺激,每种5分钟:合成肠液(SIF)、SIF+EpOME(1μM)或载体(DMSO 0.03%(v/v))、SIF+EpOME(或载体)+辣椒素(500nM),SIF。数据表示来自6个单独坐骨神经的均值±SEM。在PBS、9,10-EpOME、辣椒素或9,10-EpOME+辣椒素刺激15分钟后富含神经元的DRG培养物的iCGRP释放;a:9,10-EpOME 1μM,b:辣椒素400nM,c:9,10-EpOME 2.5μM。数据表示来自6只小鼠的DRG培养物的均值±SEM;#,*p<0.05,**p<0.01,***p<0.001学生t检验。虚线表示测定灵敏度。
图7:在紫杉醇诱导的神经病理性疼痛中CYP2J6上调。(a)野生型C57Bl/6N小鼠在注射紫杉醇(6mg·kg-1i.p.)后的机械阈值时程。bl:基线,数据表示每组10只小鼠的均值±SEM。8天后,切下坐骨神经、DRG和脊髓背角。(b)紫杉醇注射(6mg·kg-1i.p.)后8天时鼠科CYP表氧化酶转录物的表达。数据表示来自每组4只小鼠的DRG的均值±SEM;*p<0.05,**p<0.01,学生t检验。(c)在C57B16/N小鼠中i.p.注射载体(黑色)或紫杉醇(灰色,6mg·kg-1)后8天时坐骨神经、DRG、和脊髓背角中9,10-EpOME的浓度;**p<0.01,学生t检验。(d)通过LC-MS/MS分析揭示的,在紫杉醇治疗后8至9天,鼠科DRG中类二十烷酸和亚油酸代谢物合成的方案。结构从网站(lipidmaps.org)获得。
图8:特非那定对CYP2J6的抑制在体内减小了脂浓度并缓解了紫杉醇诱导的CIPNP。(a)在紫杉醇(6mg·kg-1i.p.和1mg·kg-1特非那定(灰色)或载体(2%DMSO v/v,黑色))治疗后8天时坐骨神经、DRG和脊髓背角中通过LC-MS/MS测定的9,10-EpOME的水平,显示为对照的百分比。数据表示来自每组5只小鼠的DRG的均值±SEM;*p<0.05,**p<0.01,学生t检验。(b)在特非那定施用(1mg·kg-1)后,所有测量的环氧化脂类和二氢代谢物(9,10-EpOME、12,13-EpOME、9,10-DiHOME、12,13-DiHOME和14,15-EET)在坐骨神经、DRGs、脊髓背角和血浆中的剩余浓度。(c)经紫杉醇治疗8天(6mg·kg-1i.p.)并接受了特非那定(1或2mgkg-1)或载体(DMSO 2.5or 5%(v/v))静脉内注射的小鼠的机械阈值。在注射特非那定或载体后监测机械阈值长至5h。数据表示每组8至9只小鼠的均值±SEM;#,*p<0.05,使用邦费罗尼(Bonferroni)事后检验的双因素ANOVA(*1mg kg-1,#2mg kg-1特非那定)。(d)在紫杉醇注射后8天(6mg·kg--1i.p.)并接受了氯雷他定(1mg kg-1)或载体(DMSO 2.5or 2.5%(v/v))静脉内注射的小鼠的机械阈值。数据表示每组6至9只小鼠的均值±SEM。
图9:计算的CYP2J2和CYP3A4的抑制值的相关性。位于左上象限的拮抗剂为CYP2J2选择性的。根据制造商的规程进行发光CYP2J2测定(https://www.promega.de/resources/pubhub/enotes/cytochrome-p450-2j2-enzyme-assay-using-a-novel-bioluminescent-probe-substrate/)。为了测试候选CY2J2拮抗剂的选择性,采用了特异于CYP3A4的另外的发光CYP测定,并将候选CYP2J2拮抗剂的抑制活性与该相同拮抗剂对CYP3A4的抑制活性进行比较。
SEQ ID NO:1至14:引物序列
实施例
材料与方法
动物
所有的动物实验都按照国立卫生研究院的实验动物护理和使用指南中的建议进行,并得到当地动物研究伦理委员会(Darmstadt)的批准,许可号码F95/42。对于所有的行为实验,发明人仅使用购自商业饲养公司(Charles River,Sulzfeld,Germany,Janvier,Le Geneset-Saint-Isle,FR)的6至12周龄的C57BL/6N小鼠。为了比较机械阈值,发明人采用了年龄和性别匹配的同窝小鼠作为对照。
前列腺素受体缺陷小鼠(DPI-/-、IP-/-、EP2-/-和EP4-/-)饲养在法兰克福的临床药理学研究所(Institute of Clinical Pharmocology,Frankfurt),如之前所描述的。
化疗诱导的神经病理性疼痛的紫杉醇模型
将紫杉醇溶解在1:1的聚氧乙烯蓖麻油(Cremophor EL)/乙醇中,并在盐水中稀释。腹膜内注射的剂量设置为6mg/kg,如之前所描述的。
行为测试
为了确定机械性触痛或热超敏,将小鼠保持在升高的格子上的试验笼中至少2h,以让它们适应。采用动态足底触觉计或商品名为Hargreaves的装置(Ugo Basile,Comerio,VA,Italy)进行基线测量,检测机械刺激后后爪的收缩潜伏期。对于机械阈值的评估,将钢杆推向后爪中部足底,压力线性增加(10秒内0-5g,增加0.5g/s),直至出现快速的收缩反应。不对爪的缓慢移动计数。以秒±0.1确定爪收缩潜伏期(PWL),截止时间20s。非注射和注射爪轮流测量,间隔5至10min。为了确定热阈值,将小鼠在第一天保持在升温的玻璃板(32℃)上的试验笼中至少2h,以让它们适应。接着,用高强度投射灯构成的辐射热设备刺激爪的中部足底区域,直至发生收缩。非注射和注射爪轮流测量,间隔5至10min。对于所有的行为测试,研究者都不知小鼠的治疗或基因型。
治疗:对于外周注射,将20ul的9,10-EpOME[5μM](Cayman,Ann Arbor,MI,USA)皮下(s.c.)注射进后爪的中部足底区域。对照动物接受相应体积的DMSO(Sigma,Deisenhofen,Germany;盐水中1.6%(v/v))。对于鞘内注射,通过在清醒、有意识小鼠中直接腰椎穿刺来注射3.2%DMSO/盐水(v/v)中的5ul 9,10-EpOME[10μM]。在尾静脉中静脉注射特非那定或氯雷他定(二者都来自Tocris,Bristol,UK)。
原发性背根神经节(DRG)培养
将鼠科DRG从脊髓节段切下并直接转移到含有CaC12和MgC12(Invitrogen,Carsbad,CA,USA)的冰冷HBSS中。接着,在37℃将分离的DRG在含有L-谷氨酰胺[2mM]青霉素(100U/ml)、链霉素(100μg/ml)、B-27和庆大霉素(50μg/ml)(都来自Invitrogen,Carlsbad,CA,USA)的神经基的(neurobasal)培养基中与胶原酶/分散酶(500U/ml胶原酶;2.5U/ml分散酶)温浴75min。在去除胶原酶/分散酶溶液后,用含有10%FCS的神经基的培养基清洗细胞两次,并以0.05%胰蛋白酶(Invitrogen,Carlsbad,CA,USA)温浴10min。重复清洗步骤,并用1ml吉尔森吸管将细胞机械解离。最终,将神经元置于聚L-赖氨酸(Sigma,Deisenhofen,Germany)包被的盖玻片上,并以含有L-谷氨酰胺[2mM]青霉素(100U/ml)链霉素(100μg/ml)、B-27和庆大霉素(50μg/ml)的神经基的培养基温浴过夜,直至通过钙成像评估。
钙成像实验
采用两种不同的方案来进行钙成像实验。首先,发明人采用带有10x蔡司(Achroplan)水浸物镜(Zeiss)的阿克斯科普2(Axio scope 2)正置显微镜(Zeiss,Jena,Germany)。该显微镜配备有伊梅戈(Imago)CCD相机和多色IV(Polychrome IV)单色器(都来自TILL Photonics,Grafelfmg,Germany)。在两个波长(340nm和380nm)每2秒获得图像,并采用提尔维森(Tillvision)软件23处理。随后,采用了莱卡(Leica)钙成像方案,其由配备了DFC360FX(CCD-)相机、芙拉2(Fura-2)滤镜和N-普兰(N-Plan)10x/0.25Ph1物镜的莱卡(Leica)DMI 4000b倒置显微镜(都来自Leica Microsystems,Wetzlar,Germany)构成。每2秒照相,并采用LAS AF软件处理。对于每个实验,发明人选择具有大量细胞的区域并同时监测40-110个细胞。采用制备后24-48小时的DRG神经元进行钙成像实验。让细胞加载5μM芙兰2(fura-2)-AM酯和0.02%普朗尼克(Pluronic)F-127(二者来自Biotium,Hayward,CA),并在37℃温浴30至60min。然后,以外部溶液(以mM为单位,含有:NaCl[145]、CaCl2[1.25]、MgCl2[1]、KCl[5]、D-葡萄糖[10]、HEPES[10];调整至pH 7.3)洗涤细胞。在外部溶液中以1至2ml/min的流速进行基线测量。通过去除CaC12并添加EGTA[2mM]来产生无钙溶液,通过将NaCl浓度增加至150mM来进行渗透压控制。将HC-030031(Sigma,Deisenho-fen,Germany)、AMG 9810,H89-二盐酸盐、8-溴-cAMP、GF 109203X(都来自Tocris,Bristol,UK)和NGF(Merck Millipore,Darmstadt,GE)的储备液在外部溶液中稀释至它们的最终浓度。
定量实时PCR
在指出的时间点从小鼠切下腰部DRG,并采用商品名为mirVanaTM的miRNA分离试剂盒(Ambion,life technologies,Carlsbad,CA,USA)提取RNA。采用商品名为的系统(lifetechnologies,Carlsbad,CA,USA)进行逆转录和实时PCR,并按之前所描述的用AAC(T)方法评估24,25。以下的寡核苷酸用于cDNA的扩增:
表1:用于从鼠科组织进行定量实时PCR的引物序列,a=MGH引物库,ID:160948617c2。
通过液相色谱-串联质谱法(LC-MS/MS)确定EET
样品提取和标准品:按之前所描述的方式进行样品提取。简言之,在甲醇中制备含有2500ng/ml所有分析物的储备溶液。通过进一步稀释获得工作标准品,对于EET、EpOME和DiHOME以及HODE浓度范围为0.1-250ng/ml。通过液液萃取进行样品提取。因此,用600ul乙酸乙酯将组织或细胞培养培养基萃取两次。在柔和的氮气流下,于45℃的温度去除合并的有机相。将残留物以50ul的甲醇/水(50:50,v/v)重建,10,000xg离心2min,然后在注入LC-MS/MS系统前转移到玻璃瓶(Macherey-Nagel,Duren,Germany)中。
测量环氧脂类和HODE的仪器:该LC-MS/MS系统由API 4000三重四极杆质谱仪(Applied Biosystems,Darmstadt,Germany)构成,配备有负ESI模式操作的图伯-V-源(Turbo-V-source)、安捷伦1100(Agilent 1100)双HPLC泵和脱气器(Agilent,Waldbronn,Germany)以及带有25ulLEAP注射器的HTC Pal自动加样器(Chromtech,Idstein,Germany)。用于质谱的高纯度氮通过NGM 22-LC-MS氮发生器(cmc Instruments,Eschbom,Germany)产生。对于色谱分离,使用了基迷你(Gemini)NX C18柱和前置柱(150mm x 2mm i.d.,5μM粒径和孔径,来自Phenomenex,Aschaffenburg,Germany)。采用线性梯度,流动相流速0.5ml/min,总运行时间17.5分钟。流动相A为水/氨(100:0.05,v/v),B为乙腈/氨(100:0.05,v/v)。在12min内,让梯度从85%A开始,直至10%。保持10%A 1min。在0.5min内,让流动相移回85%A并保持3.5min以平衡色谱柱,用于下一样品。样品的注射体积为20ul。以分析软件(Analyst Software)V 1.4.2(Applied Biosystems,Darmstadt,Germany)采用内标法(同位素稀释质谱法)进行定量。将分析物峰面积和内标面积的比例(y轴)对浓度(x轴)作图,通过最小二乘回归计算标准曲线,以1/浓度2加权。
[35S]GTPγS结合测定
为了测量对预测的G蛋白偶联受体的激活,采用1μM 9,10-EpOME(Cayman,Ann Arbor,MI,USA)和新制的[35S]GTPγS(1250Ci/mmol,Perkin Elmer,Waltham,MA,USA)对成年大鼠的DRG的膜制备物进行了GTPγS结合测定法。
iCGRP的测量
按之前的描述32,采用CGRP-酶免疫测定试剂盒(SpiBio,Bertin pharma,France)进行CGRP测量。对于来自DRG培养物的CGRP测量,将野生型BL/6N小鼠的DRG切下并按上文所述处理,并在48孔板中培养过夜。
数据分析和统计
所有的数据都表示均值±s.e.m。为了确定所有行为实验中的统计学显著差异,使用用于重复测量的方差分析(ANOVA),接着采用商品名为GraphPad Prism的软件进行事后邦费罗尼校正。对于仅两组比较的体外实验,进行学生t检验。P<0.05被认为是统计上显著的。
细胞色素P450荧光素酶测定
按照制造商的说明书进行CYP2J2和CYP3A4Glo测定(P450-GloTM,Promega)。
CYP2J2测定规程:
-采用商品名为MultiDrop的产品来制备CYP2J2-酶(2nM)/荧光素-2J2/4F12底物(2μM)混合物,5μl/孔,
-采用商品名为Echo的产品添加50nl/孔的化合物(10μM终浓度)/DMSO(0.5%终浓度)
-在37℃温浴30min
-采用商品名为MultiDrop的产品添加NADPH再生溶液,5μl/孔
-在37℃温浴30min
-采用商品名为MultiDrop的产品添加LDR-酯酶溶液,10μl/孔
-在37°温浴30min,在读取物(EnSpire)上读出发光值
CYP3A4测定规程:
-采用商品名为MultiDrop的产品制备CYP3A4-酶(2nM)/荧光素-IPA底物(7μM)混合物,5μl/孔,
-采用商品名为Echo的产品添加50μl/孔的化合物(10μM终浓度/DMSO(0.5%终浓度)
-在37℃温浴30min,
-采用商品名为MultiDrop的产品添加NADPH再生溶液,5ul/孔
-在37℃温浴30min,
-采用商品名为MultiDrop的产品添加LDR-酯酶溶液,10μl/孔
-在37°温浴30min,并在读取物(EnSpire)上读出发光值。
实施例1:化疗诱导的神经病理性疼痛中的CYP衍生脂质
为了研究CYP衍生脂质是否可以在化疗诱导的神经病理性疼痛中起作用,发明人将紫杉醇或载体注射进野生型BL/6N小鼠中,并在注射后24小时切下坐骨神经、DRG和脊髓背角。采用LC-MS/MS确定脂质浓度。发现是氧化的亚油酸代谢物9,10-EpOME的浓度(图1A),而不是其姊妹脂质12,13-EpOME(图1B)或他们的二氢代谢物9,10-和12,13-DiHOME(参见补充图1)在DRG中强烈升高(图1A)。还定量了9-和13-HODE的水平(图1C,1D),它们在炎性疼痛期间产生,为TRPV1的内源性激活因子33。但是,发明人不能在紫杉醇处理后检测到它们水平的任何差异。为了研究DRG中增加的9,10-EpOME浓度是否是紫杉醇处理特异性的,将酵母聚糖注射进野生型BL/6N小鼠的后爪,以诱导炎性疼痛。在注射后24h炎症高峰时,切下L4-L6-DRG和对应的背角节段。通过LC-MS/MS的脂质定量未揭示炎性疼痛期间9,10-EpOME水平的任何差异(图1E)。
接着,发明人在钙成像实验中就9,10-EpOME对DRG神经元的影响进行了表征。发明人观察到,用10μM 9,10-EpOME短刺激30s在DRG神经元中引起钙瞬变(图2A)。发明人进行了剂量反应分析以研究9,10-EpOME引发钙瞬变的效力,发现最多10.3%的DRG神经元应答25μM的9,10-EpOME,对于更高浓度,应答神经元百分比没有显著增加(图2B)。为了分析9,10-EpOME引起的钙瞬变是由细胞内钙存储的释放还是由细胞外钙的内流所导致的,发明人使用了含有2mM EGTA的无钙外部溶液,并用10μM 9,10-EpOME刺激DRG神经元两次,每次30s。在第二次刺激之前两分钟,洗入无钙外部溶液,则神经元不再应答9,10-EpOME,由此表明9,10-EpOME导致外部钙内流(图2C,2D)。用于神经元的阳性对照为50mM KCl 30s的终刺激。
为了鉴定所涉及的离子通道,使用TRPV1(AMG 9810,1μM)和TRPA1(HC-030031,20μM)的选择性拮抗剂,以便阻断9,10-EpOME导致钙内流。用9,10-EpOME(10μM,30s)刺激DRG神经元两次,并在第二次9,10-EpOME刺激之前用TRP通道拮抗剂预温浴细胞两分钟。发明人观察到,是选择性TRPV1拮抗剂AMG 9810,而不是TRPA1拮抗剂HC-030031可阻断第二次9,10-EpOME引起的钙瞬变,表明TRPV1为9,10-EpOME的靶向通道(图2E,2F)。
实施例2:9,10-EpOME敏化TRPV1
然后,发明人分析了9,10-EpOME在较低的和更生理的浓度(1μM)下是否也能够敏化TRPV1或TRPA1。由此,发明人用辣椒素刺激DRG神经元两次(200nM,15s),并在第二次辣椒素刺激前用9,10-EpOME[1μM]或载体温浴细胞两分钟,观察到经9,10-EpOME温浴的DRG神经元对辣椒素的显著强烈的应答,表明9,10-EpOME敏化TRPV1(图3A)。为了研究9,10-EpOME依赖性TRPV1敏化的效力,采用250nM至2μM的9,10-EpOME浓度进行了剂量反应分析。观察到与载体相比的第二次辣椒素应答幅度的剂量依赖性增加。这种效应看起来特异于TRPV1,因为芥末油依赖的TRPA1应答不被9,10-EPOME[1μM]敏化(图2B)。
为了用电生理手段确认9,10-EpOME对TRPV1敏化的作用,发明人采用两次辣椒素刺激[1μM]并在第二次辣椒素刺激之前用9,10-EpOME[1μM]温浴细胞,测量了来自脊髓切片的流明II(lumina II)神经元的sEPSC(图3C)。单独的9,10-EpOME处理稍稍增加sEPSC的频率。但是,与辣椒素组合时,sEPSC频率强烈增加(图3D)。然而,无论是9,10-EpOME、TRPV1,还是两种物质的组合,都没有观察到sEPSC的幅度差异(图3E)。
因为已知脂质介导的TRPV1敏化大多数涉及到G蛋白偶联受体的激活,发明人进行了GTPγS测定,以分析9,10-EpOME是否能够在DRG中激活GPCR,并在用1μM 9,10-EpOME温浴后观察到显著增加的GTPγS信号(图4A)。为了鉴定9,10-EpOME介导的TRPV1敏化的机制,发明人接着测量了经载体、9,10-EpOME、IP受体激动剂西卡前列素或毛喉素[每种1μM]刺激15分钟的富含神经元的DRG培养物中的cAMP。有趣的是,发明人观察到,与其载体相比,9,10-EpOME引起了cAMP浓度的显著增加(图4B)。这些结果表明了9,10-EpOME对盖尔菲斯(Galphas)偶联受体的激活。
由于TRPV1可被PKA和PKC磷酸化,二者导致该通道增加的活性和敏化35,所以发明人在钙成像实验中用培养的来自野生型BL/6N小鼠的DRG神经元研究了PKA或PKC抑制剂是否可减弱9,10-EpOME引起的TRPV1敏化。发明人采用与上文提到的相同规程(两次辣椒素刺激,之间用9,10-EpOME温浴)可重复辣椒素依赖的TRPV1敏化。但是,发明人观察到,用PKA-抑制剂(H89二盐酸盐,10μM,1h)预温浴导致9,10-EpOME引起的TRPV1敏化的显著减弱(图4C,4D)。在相同条件下(10μM,预温浴1h)使用PKC-抑制剂GF 109203X(GFX)对9,10-EpOME引起的TRPV1敏化无任何影响(图4E,4F),由此表明9,10-EpOME引起PKA-而不是PKC-介导的TRPV1敏化。
发明人接着在钙成像实验中测试了盖尔菲斯(Galphas)偶联的前列腺素受体参与9,10-EpOME依赖的TRPV1敏化的可能性。前列腺素受体对它们的配体前列腺素类具有不同的特异性,也可能由其它的脂质激活。但是,发明人未能在前列腺素E受体EP2和EP4或者前列腺素D或I受体(DP和IP受体)缺陷小鼠中观察到9,10-EpOME引起的TRPV1敏化的任何减弱。为了表征9,10-EpOME的体内作用,发明人将该脂质注射进野生型BL/6N小鼠的后爪内,并测量热(图5A)和机械阈值(图5B),直至注射后5h。两种情况下,9,10-EpOME引起疼痛阈值的显著降低,在注射后持续1h(热)或2h(机械)(图1A,1B)。然后,发明人将9,10-EpOME鞘内注射,并在短时间间隔内测量热和机械阈值。在鞘内注射后30分钟观察到显著但相当弱的热阈值降低(图5C)。但是,在9,10-EpOME鞘内注射后长至1.5h观察到机械阈值降低(图5D)。
由于TRPV1升高的活性导致促进神经源性炎症的降钙素基因相关肽(CGRP)的释放增加37,所以发明人分析9,10-EpOME是否能够增加TRPV1依赖的CGRP释放。发明人从野生型BL/6N小鼠切下坐骨神经,并将它们与单独的9,10-EpOME[1μM]、或者连同辣椒素[500nM]一起温浴,对于辣椒素和9,10-EpOME的共刺激观察到CGRP释放的强烈增加。CGRP释放显著高于仅采用辣椒素或9,10-EpOME(图6A)。为了研究在细胞胞体中是否也可见这种作用,用9,10-EpOME、辣椒素或这两物质刺激富含神经元的DRG培养物,采用两种不同的EpOME浓度[1和2.5μM]。同样,采用EpOME和辣椒素二者,与采用任一种物质相比,iCGRP的释放显著增加。但是,采用2.5μM 9,10-EPOME,没有CGRP释放的显著增加(图6B)。
实施例3:CYP2J2调节9,10-EpOME
接着,研究了9,10-EpOME合成在紫杉醇CIPNP期间是如何被调节的。因为9,10-EpOME被认为是由亚族2C和2J的CYP表氧化酶所合成的16,38,发明人考察了这些亚族的鼠科CYP表氧化酶的表达。在紫杉醇处理后第8天,发明人在紫杉醇处理小鼠的机械阈值中观察到稳定的平台(图7A)。
然后发明人切下载体和紫杉醇处理小鼠的DRG,并研究了鼠科CYP2C29、CYP2C37、CYP2C38、CYP2C39、CYP2C44、CYP2J6和CYP3A11的表达。但是,在鼠科DRG中未能检测到CYP亚型2C29和2C44。发明人观察到,与载体处理相比,在紫杉醇处理小鼠的DRG中CYP2J6显示出最强表达(图7B)。紫杉醇处理后第8天时,CYP2J6的这种增加的表达与9,10-EpOME的增加的水平相关,如通过LC-MS/MS测量坐骨神经、腰DRG和脊髓所分析的。
实施例4:CYP2J2拮抗剂抑制9,10-EpOME合成并减轻CIPNP
将人CYP2J2(鼠科CYP2J6的同源蛋白)的强力抑制剂特非那定用作拮抗剂。因为对特非那定与人CYP2J2的作用位点已有描述,所以发明人将鼠科CYP2J6和人CYP2J2的氨基酸进行比对,并在两种蛋白的相同位置发现了所有推测的与特非那定相互作用的位点(Leu83、Met116、Ile127、Phe30、Thr315、Ile376、Leu378、Val380、Leu402和Thr488),除了替换为谷氨酰胺的Arg117。基于CYP2J2和CYP2J6之间惊人的氨基酸序列相似性,特非那定也与CYP2J6相互作用,并抑制该蛋白。为了研究特非那定对脂质水平的影响,发明人向8天前接受了紫杉醇的小鼠静脉注射1mg·kg-1特非那定。两小时后,发明人切下坐骨神经、DRG和背侧脊髓,并在这些组织中定量环氧化脂类。发明人可在所有研究的组织中观察到9,10-EpOME浓度的显著降低(图8A)。发明人还观察到,所有测量的环氧化脂类和它们的(9,10-EpOME、12,13-EpOME、9,10-DiHOME、12,13-DiHOME和14,15-EET)的剩余浓度在DRG、脊髓背角和血浆中都有显著降低,但在特非那定处理的动物的坐骨神经中并非如此(图8B)。
发明人接着在小鼠中研究了特非那定处理是否可以减轻紫杉醇诱导的CIPNP。因此,发明人将特非那定(1或2mg·kg-1或载体(DMSO))静脉注射进8天前接受了紫杉醇的小鼠中。
发明人在特非那定注射后的第1、2、4和5h测量了机械阈值,可观察到经特非那定处理的小鼠的机械阈值的显著升高,持续2h。但是,未观察到两剂量之间的显著差异(图8C)。由于特非那定为组胺-1-受体拮抗剂,发明人使用了另一种不抑制CYP2J2的H1受体拮抗剂氯雷他定来研究该抗伤害作用实际上是由CYP2J2、还是组胺-1-受体的抑制引起。但是,与载体相比,氯雷他定处理不减轻紫杉醇诱导的CIPNP(图8D)。
实施例5:新的选择性CYP2J2拮抗剂的筛选
对商品名为的物质(FDA Approved Drug Library v2)筛选CYP2J2的新的选择性拮抗剂,用于本文描述的发明的情形。将基于酶催化的CYP-Glo荧光素酶的反应用于测定CYP2J2的活性,将CYP3A4作为非选择性对照。在实验中将特非那定用作阳性对照。两种筛选的结果显示在图9中。显示出对CYP2J2的抑制超过60%并且对CYP3A4的抑制约为0%的拮抗剂被认为是选择性CYP2J2拮抗剂,可用于本文描述的方法和用途中,将它们列在以下的表2中:
表2:
讨论
9,10-EpOME能够通过cAMP-PKA依赖机制在亚微摩尔浓度敏化DRG神经元中的TRPV1,导致随后的iCGRP从DRG释放。其它的氧化亚油酸代谢物(OLAM),例如9和13-HODE(它们在皮肤过度加热期间产生),已证实为直接的TRPV1激动剂并促进炎性痛觉过敏。发明人也可在鼠科组织中检测到9-和13-HODE,最主要在外周组织中。
发明人采用CYP2J2抑制剂特非那定降低9,10-EpOME的合成,可降低环氧化脂类的水平至约50%。在紫杉醇CIPNP期间,特非那定处理导致小鼠中减弱的机械超敏。CYP2J2及其同源物的拮抗剂因此可用于治疗或预防CIPNP,这是被证实的,因为经氯雷他定(选择性H1受体拮抗剂,不影响CYP2J2)处理的动物未在紫杉醇CIPNP中显示出改善,这表明所观察到的特非那定作用是由于对CYP2J2而不是组胺-1-受体的抑制。
化疗诱导的神经病理性疼痛和随后的感觉障碍仍然为细胞抑制剂的最严重副作用。尤其是在紫杉醇处理中,可观察到早期急性疼痛综合征,这看起来是由对伤害感受神经元的敏化所介导。但是,尚不知晓促进这种病理学生理状态的内源性介导物。根据发明人的数据,9,10-EpOME依赖的TRPV1敏化和伤害感受神经元的增加的活性可能由此促进了紫杉醇急性疼痛综合征(P-APS)。
目前,对于CIPNP治疗剂有强烈的未满足的医疗需求。在大型随机和安慰剂对照临床试验中,用诸如阿米福汀或谷胱甘肽的抗氧化剂或神经保护性物质治疗患者未能缓解CIPNP,最近的Cochrane综述得出结论,没有这些物质用于功能性CIPNP疗法的证据。此外,抗氧化剂可以干扰细胞抑制剂的抗肿瘤效果。最近,有报道称,用N-乙酰半胱氨酸(NAC)和维生素E处理通过减少DNA损伤而增加小鼠中肺肿瘤细胞增殖和肿瘤生长。就这点而言,CYP2J2抑制剂可能优于采用抗氧化剂,因为已有报道,它们通过激活半胱天冬酶-3、Bax和Bcl-2以及通过减少肿瘤细胞迁移和粘附,甚至在体外和体内减少肿瘤生长。
序列表
<110> 弗劳恩霍夫应用研究促进协会
<120> 疼痛治疗中的CYP2J2拮抗剂
<130> F30742WO
<150> EP14181086.1
<151> 2014-08-14
<150> EP14186624.4
<151> 2014-09-26
<160> 14
<170> PatentIn version 3.5
<210> 1
<211> 19
<212> DNA
<213> Artificial
<220>
<223> Primer sequence
<400> 1
gcctcaaagc ctactgtca 19
<210> 2
<211> 19
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 2
aacgccaaaa cctttaatc 19
<210> 3
<211> 20
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 3
atactctata tttgggcagg 20
<210> 4
<211> 18
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 4
gttcctccac aaggcaac 18
<210> 5
<211> 19
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 5
ttgccttctg taatccccc 19
<210> 6
<211> 22
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 6
tctaacgcag gaatggataa ac 22
<210> 7
<211> 17
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 7
ggagacagag ctgtggc 17
<210> 8
<211> 20
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 8
taaaaacaat gccaaggccg 20
<210> 9
<211> 20
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 9
ctttccaacg agcgattccc 20
<210> 10
<211> 22
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 10
tgtttctcct cctcgatctt gc 22
<210> 11
<211> 19
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 11
ggcctcccac ctagtggaa 19
<210> 12
<211> 22
<212> DNA
<213> Primer sequence
<400> 12
ataacctcgt ccagtaacct ca 22
<210> 13
<211> 21
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 13
gacaaacaag cagggatgga c 21
<210> 14
<211> 21
<212> DNA
<213> artificial
<220>
<223> Primer sequence
<400> 14
ccaagctgat tgctaggagc a 21
- 还没有人留言评论。精彩留言会获得点赞!