HER2抗体‑药物缀合物的制作方法
本申请根据35U.S.C.§119(e)要求2014年6月23日提交的美国临时专利申请U.S.S.N.62/015,661和2014年6月20日提交的U.S.S.N.62/014,912的优先权,其各自在此引入作为参考。
发明领域
本发明属于抗癌治疗的领域,且通过抗体-药物缀合物(ADC)形式,尤其对癌细胞,提供细胞毒性药物递送的功效和特异性。
背景技术:
抗体-药物缀合物(ADC)为将单克隆抗体(mAbs)的特异性与细胞毒性分子的效能组合的一类治疗剂。ADC的使用通过缀合的细胞毒性剂而赋予抗体癌症杀伤活性,同时靶特异性递送避免由暴露于游离毒性剂而造成的全身毒性。目前,两种ADC已经FDA批准而用于治疗人类癌症。(Brentuximab vedotin或SGN-35),一种与细胞毒性剂MMAE缀合的抗CD30抗体,设计用于治疗CD30-阳性复发性淋巴瘤。(T-DM1),一种与细胞毒性剂DM1缀合的抗-HER2抗体,设计用于治疗HER2-阳性转移性乳腺癌。
连接基技术深刻影响ADC效力、特异性和安全性。酶不稳定连接基利用细胞内部和外部的蛋白酶的差别化活性以实现对药物释放的控制。药物可以通过肽键与抗体缀合,并且仅可以通过存在于细胞内的溶酶体蛋白酶的作用特异性断裂,并且在某些肿瘤类型中以升高的水平被特异性断裂(Koblinsk等人,2000)。这将确保连接基在血流中的稳定性,以限制对健康组织的损伤。然而,一些酶不稳定的连接基的增加的相关疏水性可导致ADC的聚集,特别是对于强疏水性药物。因此,需要能够提供血清稳定性以及增加的溶解性的连接基,允许疏水性药物的有效缀合和细胞内递送。
人表皮生长因子受体2蛋白,HER2(ErbB2)是表皮生长因子受体家族的成员。已知这些受体酪氨酸激酶在发育和肿瘤发生中都起关键作用。在25%-30%的原发性乳腺癌中观察到HER2蛋白的过表达(Press等人,1993),因此成为癌靶向治疗的重要候选物。已经在体外和在小鼠异种移植模型中显示人源化抗HER2抗体曲妥珠单抗抑制过表达HER2的人肿瘤细胞的增殖(Hudziak等人,1989;Baselga等人,1998)。虽然曲妥珠单抗是临床上有活性的并且在治疗患有HER2过表达转移性乳腺癌的患者中有效(Baselga等,1996),但最初对曲妥珠单抗应答的大多数人在一年内产生抗性(Romond等,2005;Nahta等,2006;Pohlmann等,2009)。因此,需要开发针对过表达HER2的肿瘤细胞的新疗法。
技术实现要素:
本发明的化合物包含药物部分、能靶向所选择的细胞群的靶向部分、和包含酰基单元的连接基、用于提供在药物部分和靶向部分之间的距离的任选的间隔基单元、可在合适的条件下断裂的肽连接基、亲水性自降解(self-immolative)连接基、和任选的第二自降解间隔基或环化自消除型连接基。
本发明还提供式(I)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
X为亲水性自降解连接基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
本发明还提供式(II)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
在一些实施方案中,提供式(Ia)的化合物:
或其盐或溶剂合物或立体异构体;其中D、T、X、L1、L2、L3、L4和A如式(I)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些实施方案中,提供式(IIa)的化合物:
或其盐或溶剂合物或立体异构体;其中D、T、L1、L2、L3、L4和A如式(II)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在式I、II、Ia和IIa的化合物的一些实施方案中,T为抗体靶向分子。在一些实施方案中,T为抗HER2抗体。在一些实施方案中,T为单克隆抗HER2抗体。在一些实施方案中所述单克隆抗HER2抗体为帕妥珠单抗。在一些实施方案中,所述单克隆抗HER2抗体为margetuximab。在一些实施方案中,T为人源化抗HER2抗体。在一些实施方案中,所述人源化抗HER2抗体为曲妥珠单抗。
在式I、II、Ia和IIa的化合物的一些实施方案中,该抗体的重链和/或轻链的一个或多个氨基酸残基被半胱氨酸残基代替。在一些实施方案中,该抗体的Fc区的一个或多个氨基酸残基被半胱氨酸残基代替。在一些实施方案中,该抗体的一个或多个氨基酸残基在轻链的位置147、188、200、201和/或206,和/或在该重链的位置155、157、165、169、197、199、209、211和/或442被半胱氨酸残基代替,使用EU编号(Kabat中的EU索引)。在一些实施方案中,D通过半胱氨酸残基连接至T。在一些实施方案中,D为含氨基的药物部分,其中该药物通过含氨基的药物部分的氨基连接至L1或X。在一些实施方案中,D为多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或丝裂霉素C(MMC),或它们的氨基衍生物。
在上述实施方案任一个中,A-L4-L3-L2-X-L1-D为:
本发明一些方面涉及药物组合物,其包含本文所述的化合物或其盐或溶剂合物或立体异构体;和药学上可接受的载体。
本发明一些方面涉及杀死细胞的方法。该方法包括向细胞给药足以杀死细胞的量的本文所述的化合物或其盐或溶剂合物或立体异构体或药物组合物。在一些实施方案中,所述细胞为癌细胞。在一些实施方案中,所述癌细胞为乳腺癌细胞、胃癌细胞或卵巢癌细胞。
本发明一些方面涉及在需要的个体中治疗癌症的方法。该方法包括向个体给药有效量的本文所述的化合物或其盐或溶剂合物或立体异构体或药物组合物。在一些实施方案中,所述癌症为乳腺癌、胃癌或卵巢癌。
本发明一些方面涉及式I、Ia、II或Iia的化合物或其盐或溶剂合物或立体异构体或药物组合物,其用于治疗癌症。在一些实施方案中,所述癌症为乳腺癌、胃癌或卵巢癌。
在一些实施方案中,L1为键。在一些实施方案中,L1为自降解连接基。在一些实施方案中,L1为氨基苄基氧基羰基连接基。在一些实施方案中,
L1选自
其中n为1或2。
在一些实施方案中,L1选自
在一些实施方案中,L2为键。在一些实施方案中,L2为自降解连接基。在一些实施方案中,L2为氨基苄基氧基羰基连接基。在一些实施方案中,L2选自
其中n为1或2。
在一些实施方案中,L3为具有1至10个氨基酸残基的肽连接基。在一些实施方案中,L3为具有2至4个氨基酸残基的肽连接基。在一些实施方案中,L3为具有1、2、3、4、5、6、7、8、9或10个氨基酸残基的肽连接基。在一些实施方案中,L3为包含至少一个赖氨酸或至少一个精氨酸残基的肽连接基。在一些实施方案中,L3为包含选自以下的氨基酸残基的肽连接基:赖氨酸、D-赖氨酸、瓜氨酸、精氨酸、脯氨酸、组氨酸、鸟氨酸和谷氨酰胺。在一些实施方案中,L3为包含选自以下的氨基酸残基的肽连接基:缬氨酸、异亮氨酸、苯丙氨酸、蛋氨酸、天冬酰胺、脯氨酸、丙氨酸、亮氨酸、色氨酸和酪氨酸。在一些实施方案中,L3为选自以下的二肽单元:缬氨酸-瓜氨酸、脯氨酸-赖氨酸、蛋氨酸-D-赖氨酸、天冬酰胺-D-赖氨酸、异亮氨酸-脯氨酸、苯丙氨酸-赖氨酸和缬氨酸-赖氨酸。在一些实施方案中,L3为缬氨酸-瓜氨酸。
在一些实施方案中,L4为键。在一些实施方案中,L4为间隔基。在一些实施方案中,所述间隔基为聚亚烷基二醇、亚烷基、亚烯基、亚炔基、或聚胺。在一些实施方案中,L4为L4a-C(O),L4a-C(O)-NH,L4a-S(O)2,或L4a-S(O)2-NH,其中各L4a独立地为聚亚烷基二醇、亚烷基、亚烯基、亚炔基、或聚胺。在一些实施方案中,L4为L4a-C(O),其中L4a为聚亚烷基二醇、亚烷基、亚烯基、亚炔基、或聚胺。在一些实施方案中,L4为L4a-C(O),其中L4a为聚亚烷基二醇。在一些实施方案中,L4为L4a-C(O),其中L4a为聚乙二醇。在一些实施方案中,所述间隔基为式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m为0至30的整数。在一些实施方案中,所述间隔基为式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m为整数1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30。在一些实施方案中,L4为L4a-C(O),其中L4a为亚烷基。
在一些实施方案中,A选自
其中各Q2为NH或O,且各q为整数1至10,且各q1独立地为1至10的整数。在一些实施方案中,q为2、3、4或5。在某些实施方案中,q1为2、3、4或5。在其它实施方案中,各q为整数1、2、3、4、5、6、7、8、9或10。在一些实施方案中,A为
其中各Q2独立地为NH或O且各q独立地为1至10的整数。在其它实施方案中,各q独立地为整数1、2、3、4、5、6、7、8、9或10。在一些实施方案中,q为2、3、4或5。在一些实施方案中,A选自基团
其中各Q2独立地为NH或O。
在一些实施方案中,D为含氨基的药物部分,其中该药物通过含氨基的药物部分的氨基连接至L1或X。在一些实施方案中,D为选自以下的多卡霉素的氨基衍生物:
在一些实施方案中,D为多拉司他汀的氨基衍生物(例如单甲基多拉司他汀10):
在一些实施方案中,A-L4-L3-L2为
在一些实施方案中,A-L4-L3-L2-X-L1-D为:
在一些实施方案中,A-L4-L3-L2-X-L1-D为:
在一些实施方案中,A-L4-L3-L2-X-L1-D为:
本发明一些方面涉及制备式(II)的化合物或其盐或溶剂合物或立体异构体的方法:
其中:
D为药物部分;
T为抗体;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。该方法包括将抗体与化合物Z或其盐或溶剂合物或立体异构体反应:
本发明一些方面涉及制备式(IIa)的化合物或其盐或溶剂合物或立体异构体的方法:
其中:
p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
D为药物部分;
T为抗体;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。该方法包括将抗体与化合物Z或其盐或溶剂合物或立体异构体反应:
在一些实施方案中,该抗体为抗HER2抗体。在一些实施方案中,该抗体为单克隆抗HER2抗体。在一些实施方案中,该抗体为人源化抗HER2抗体,任选为单克隆人源化抗HER2抗体。在一些实施方案中,该抗体重链和/或轻链的一个或多个氨基酸残基被半胱氨酸残基代替。在一些实施方案中,该抗体的Fc区的一个或多个氨基酸残基被半胱氨酸残基代替。在一些实施方案中,在轻链的位置147、188、200、201和/或206,和/或在重链的位置155、157、165、169、197、199、209、211和/或442,该抗体的一个或多个氨基酸残基被半胱氨酸残基代替,使用EU编号(Kabat中的EU索引)。在一些实施方案中,该抗体包括一个或多个巯基。在一些实施方案中,该化合物使用本文所述的方法之一制备,其中所述抗体包括一个或多个巯基。
还提供了药物组合物,其包含本文所述的化合物或其盐或溶剂合物或立体异构体,和药学上可接受的载体。
本发明一些方面涉及式(IX)的化合物
或其盐或溶剂合物或立体异构体;其中R为NO2或NH2.
本发明一些方面涉及制备化合物X或其盐或溶剂合物或立体异构体的方法:
其中:
L2为键或自降解连接基;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。该方法包括将化合物W:A-L4-L3-L2;与化合物I反应:
本发明一些方面涉及制备化合物Z或其盐或溶剂合物或立体异构体的方法:
其中:
D为药物部分;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
该方法包括:将化合物X:和氯甲酸对硝基苯酯反应以形成化合物Y:
和将化合物Y与包含L1-D的化合物反应。
本发明一些方面涉及制备化合物X1或其盐或溶剂合物或立体异构体的方法:
其中:
L2为键或自降解连接基;
L3为肽连接基;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。该方法包括:将化合物W1:L3-L2;和化合物I反应:
本发明一些方面涉及制备化合物Y1或其盐或溶剂合物或立体异构体的方法:
其中:
D为药物部分;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。该方法包括:在氯甲酸对硝基苯酯的存在下将化合物X1:
和包含L1-D的化合物反应。在一些实施方案中,在制备化合物Y1的方法中,将选自二(4/对硝基苯基)碳酸酯、光气、三光气/双(三氯甲基碳酸酯)、氯甲酸三氯甲酯、N,N’-二琥珀酰亚胺基碳酸酯和1,1’-羰基二咪唑的化合物,代替氯甲酸对硝基苯酯。
本发明一些方面涉及制备化合物Z或其盐或溶剂合物或立体异构体的方法:
其中:
D为药物部分;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;
A为酰基单元;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
该方法包括:将化合物Y1:
和包含A-L4的化合物反应。
还提供了下式的化合物:
或其盐或溶剂合物或立体异构体;
其中:
L2为键或自降解连接基;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
还提供了下式的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
还提供了下式的化合物:
或其盐或溶剂合物或立体异构体;
其中:
L2为键或自降解连接基;
L3为肽连接基;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
还提供了下式的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;且
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
本发明还提供式(III)的化合物:
或其盐或溶剂合物或立体异构体;NH
其中T为靶向部分。
在一些实施方案中,提供了式(IIIa)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(IV)的化合物:
或其盐或溶剂合物或立体异构体;
其中T为靶向部分。
在一些实施方案中,提供了式(IVa)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(V)的化合物:
或其盐或溶剂合物或立体异构体;
其中T为靶向部分。
在一些实施方案中,提供了式(Va)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(VI)的化合物:
或其盐或溶剂合物。
本发明提供式(VII)的化合物:
或其盐或溶剂合物。
本发明提供式(VIII)的化合物:
本发明提供式(IX)的化合物:
或其盐或溶剂合物或立体异构体;其中R为NO2或NH2。
在某些实施方案中,式(I)、(II)、(III)、(IV)、(V)、(VI)、(VII)、(VIII)或(IX)的化合物为选自本文发明详述中描述或示例的那些物质的化合物。
在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物的某些实施方案中,T为抗体靶向分子。在一些其它实施方案中,T为抗HER2抗体。在其它实施方案中,该抗HER2抗体为人源化或单克隆或人源化单克隆抗体。在一些实施方案中,T为人源化单克隆抗HER2抗体曲妥珠单抗。在一些实施方案中该单克隆抗HER2抗体为帕妥珠单抗。在一些实施方案中,该单克隆抗HER2抗体为margetuximab。
在其它实施方案中,该抗体的重链和/或轻链的一个或多个氨基酸残基被半胱氨酸残基代替(例如,工程化以在不存在于母体抗体中的位置包含半胱氨酸残基)。在一些实施方案中,该抗体的Fc区的一个或多个氨基酸残基被半胱氨酸残基代替。在一些实施方案中,该抗体的一个或多个氨基酸残基在该轻链的位置147、188、200、201和/或206,和/或在该重链的位置155、157、165、169、197、199、209、211和/或442,使用EU编号(Kabat中的EU索引)。在一些实施方案中,包含工程化半胱氨酸残基的抗体为抗HER2抗体。在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物的一些实施方案中,D通过半胱氨酸(例如工程化的)残基连接至T。
在某些实施方案中,D为含氨基的药物部分。在一些实施方案中,D通过氨基连接至L1或X。在其它实施方案中,D为多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或丝裂霉素C(MMC),或它们的氨基衍生物。
在另一方面,本发明提供药物组合物,其包含至少一种式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐。根据该实施方案的药物组合物还可包含药物可接受的赋形剂。本发明还提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐,其用作药物。
在另一方面,本发明提供杀死细胞的方法,包括向细胞给药足以杀死细胞的量的式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物。在一些实施方案中,所述细胞为癌细胞。在其它实施方案中,所述癌细胞为乳腺癌细胞、胃癌细胞或卵巢癌细胞。
在另一方面,本发明提供在需要的个体中治疗癌症的方法,包括向该个体给药有效量的式(I)-(V)或(Ia)-(Va)的化合物或其盐、溶剂合物或立体异构体。可用本文所述的方法治疗的癌症的实例包括,但不限于,乳腺、膀胱、胰腺、非小细胞非癌(NSCLC)、卵巢、子宫内膜、结肠、肾、头与颈、胃、食管、前列腺、和睾丸生殖细胞的癌症,子宫癌,维尔姆斯肿瘤。在一些实施方案中,在式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物中,T为抗HER2抗体且D为含氨基的药物部分。在其它实施方案中,T为抗体曲妥珠单抗且D为单甲基多拉司他汀10。在一些实施方案中,T为抗体帕妥珠单抗且D为单甲基多拉司他汀10。在一些实施方案中,T为抗体margetuximab且D为单甲基多拉司他汀10。
在另一方面,本发明提供式(I)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
X为亲水性自降解连接基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元,
其用于治疗癌症。
在另一方面,本发明提供式(II)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元,
其用于治疗癌症。
在另一方面,本发明提供式(II)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元,
其用于治疗癌症。
在另一方面,本发明提供式(Ia)的化合物:
或其盐或溶剂合物或立体异构体;其中D、T、X、L1、L2、L3、L4和A如式(I)所定义,且p为1至20,其用于治疗癌症。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些实施方案中,提供式(IIa)的化合物:
或其盐或溶剂合物或立体异构体;其中D、T、L1、L2、L3、L4和A如式(II)所定义,且p为1至20,其用于治疗癌症。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些方面,本发明还提供式(III)的化合物:
或其盐或溶剂合物或立体异构体;
其中T为靶向部分,其用于治疗癌症。
在一些实施方案中,提供了式(IIIa)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20,其用于治疗癌症。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(IV)的化合物:
或其盐或溶剂合物或立体异构体;
其中T为靶向部分,其用于治疗癌症。
在一些实施方案中,提供了式(IVa)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20,其用于治疗癌症。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(V)的化合物:
或其盐或溶剂合物或立体异构体;
其中T为靶向部分,其用于治疗癌症。
在一些实施方案中,提供了式(Va)的化合物:
或其盐或溶剂合物或立体异构体;其中T为靶向部分且p为1至20,其用于治疗癌症。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在另一方面,本发明提供药物组合物,其包含至少一种式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐,其用于治疗癌症。根据该实施方案的药物组合物还可包含药物可接受的赋形剂。本发明还提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐,其用作药物。
在另一方面,本发明提供式(I)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中:
D为药物部分;
T为靶向部分;
X为亲水性自降解连接基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
在另一方面,本发明提供式(II)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
在另一方面,本发明提供式(II)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
在另一方面,本发明提供式(Ia)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中D、T、X、L1、L2、L3、L4和A如式(I)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些实施方案中,提供式(IIa)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中D、T、L1、L2、L3、L4和A如式(II)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些方面,本发明还提供式(III)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分。
在一些实施方案中,提供式(IIIa)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(IV)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分。
在一些实施方案中,提供式(IVa)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明提供式(V)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分。
在一些实施方案中,提供式(Va)的化合物或其盐或溶剂合物或立体异构体在制备用于治疗癌症的药物中的用途:
其中T为靶向部分且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
在一些方面,本发明提供药物组合物(其包含至少一种式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐)在制备用于治疗癌症的药物中的用途。根据该实施方案的药物组合物还可包含药物可接受的赋形剂。本发明还提供式(I)、(II)、(III)、(IV)或(V)或(Ia)、(IIa)、(IIIa)、(IVa)或(Va)的化合物或其药物可接受的盐,其用作药物。
在以上讨论的任一实施方案中,所述抗HER2抗体包含重链可变区和轻链可变区,其中
(1)该重链可变区包含SEQ ID NO:16-18的氨基酸序列的三个重链CDR和/或该轻链可变区包含SEQ ID NO:19-21的氨基酸序列的三个轻链CDR;
(2)该重链可变区包含SEQ ID NO:22-24的氨基酸序列的三个重链CDR和/或该轻链可变区包含SEQ ID NO:25-27的氨基酸序列的三个轻链CDR;或
(3)该重链可变区包含SEQ ID NO:28-30的氨基酸序列的三个重链CDR和/或该轻链可变区包含SEQ ID NO:31-33的氨基酸序列的三个轻链CDR。
在以上讨论的任一实施方案中,该抗HER2抗体包含重链可变区和轻链可变区,其中
(1)该重链可变区包含SEQ ID NO:8的氨基酸序列和/或该轻链可变区包含SEQ ID NO:7的氨基酸序列;
(2)该重链可变区包含SEQ ID NO:13的氨基酸序列和/或该轻链可变区包含SEQ ID NO:12的氨基酸序列;
(3)该重链可变区包含SEQ ID NO:15的氨基酸序列和/或该轻链可变区包含SEQ ID NO:14的氨基酸序列。
本发明的其它实施方案、特征和优点根据以下发明详述和通过本发明的实践将是明显的。
附图简述
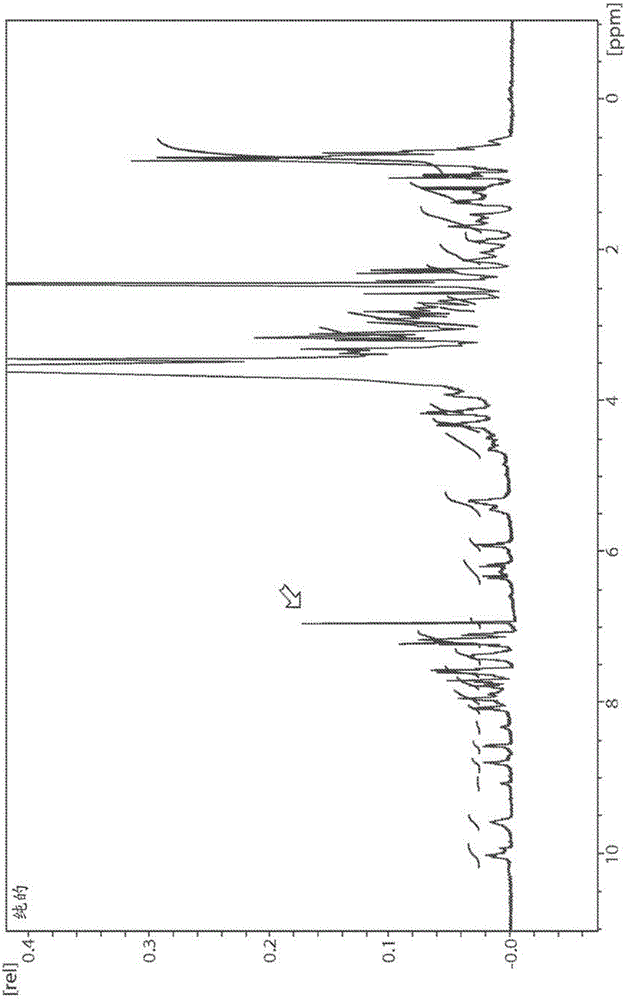
图1显示Tap-18H的NMR谱。
图2显示Tap-18Hr1的NMR谱。
图3显示Tap-18Hr2的NMR谱。
图4显示抗HER2-IgG1/TAP18Hr1对抗卵巢癌SKOV-3的体内抗肿瘤活性。
图5显示抗HER2-IgG1/TAP18Hr1对抗乳腺癌MDA-MB-453的体内抗肿瘤活性。箭头指示ADC治疗的时间(第1天)。
图6显示抗HER2-IgG1/TAP18Hr1对抗胃癌NCI-N87的体内抗肿瘤活性。箭头指示ADC治疗的时间(第1天和第22天)。
图7显示位点特异的缀合的抗HER2-Cys变体对抗胃癌NCI-N87的体内抗肿瘤活性。箭头指示ADC治疗的时间(第1天)。
图8显示Tap18Hr1常规缀合的抗HER2和位点特异的缀合的抗HER2-Cys变体对抗乳腺癌JIMT-1的体内抗肿瘤活性。箭头指示ADC治疗的时间(第1天)。
发明详述
本公开提供了具有亲水性自降解连接基的化合物,其在合适的条件下是可断裂的并且引入亲水基团以提供化合物的更好的溶解性。对于通常为疏水的细胞毒性药物,该亲水性自降解连接基可提供药物缀合物增加的溶解性。在药物缀合物中使用亲水性自降解连接基的其他优点包括药物缀合物增加的稳定性和药物缀合物降低的聚集。
本公开提供了可以具有优异的血清稳定性的药物缀合物。例如,与其中药物的羟基通过易于在水性缓冲液或人血清中快速水解的不稳定碳酸酯键连接到间隔基的药物缀合物相反,本申请的药物缀合物利用苄氧基羰基键。这些缀合物在相同条件下相对更稳定,并且在用蛋白酶例如组织蛋白酶B处理时选择性地经历断裂以释放药物。对于药物缀合物,血清稳定性是期望的性质,其中期望将惰性药物施用于患者的血清,通过配体使惰性药物集中于靶标上,然后使药物缀合物仅在靶标附近转化为活性形式。
本公开提供了可以具有减少的聚集的药物缀合物。一些酶不稳定连接基的增加的相关疏水性可能导致药物缀合物的聚集,特别是对于强疏水性药物。通过将亲水基团引入连接基中,药物缀合物的聚集减少。与具有化学不稳定连接基和不可断裂连接基的ADC相比,本文描述的连接基可以通过特异性酶不稳定设计获得更好的血清稳定性,以及通过对异质癌细胞的旁观者效应(bystander effect)实现更好的功效。本发明的化合物包含药物部分、能靶向所选择的细胞群的靶向部分、和包含酰基单元的连接基、用于提供在药物部分和靶向部分之间的距离的任选的间隔基单元、可在合适的条件下断裂的肽连接基、亲水性自降解连接基、和任选的第二自降解间隔基或环化自消除型连接基。下面讨论每个特征。
“烷基”是指具有1、2、3、4、5、6、7、8、9或10碳原子且优选1、2、3、4、5或6个碳原子的一价饱和脂族烃基。该术语包括,作为实例,直链和支链烃基如甲基(CH3-)、乙基(CH3CH2-)、正丙基(CH3CH2CH2-)、异丙基((CH3)2CH-)、正丁基(CH3CH2CH2CH2-)、异丁基((CH3)2CHCH2-)、仲丁基((CH3)(CH3CH2)CH-)、叔丁基((CH3)3C-)、正戊基(CH3CH2CH2CH2CH2-)、新戊基((CH3)3CCH2-)和正己基(CH3(CH2)5-)。
“亚烷基”是指优选具有1、2、3、4、5、6、7、8、9或10且更优选1、2或3个碳原子的二价脂族亚烃基,其为直链或支链的。该术语包括,作为实例,亚甲基(-CH2-)、亚乙基(-CH2CH2-)、正亚丙基(-CH2CH2CH2-)、异亚丙基(-CH2CH(CH3)-)、(-C(CH3)2CH2CH2-)、(-C(CH3)2CH2C(O)-)、(-C(CH3)2CH2C(O)NH-)、(-CH(CH3)CH2-),等。
“烯基”是指具有2、3、4、5、6、7、8、9或10个碳原子且优选2、3或4个碳原子和具有至少1个且优选1至2个双键位点的直链或支链烃基。该术语包括,作为实例,二-乙烯基、烯丙基和丁-3-烯-1-基。该术语中包括顺式和反式异构体或这些异构体的混合物。
“亚烯基”是指具有2、3、4、5、6、7、8、9或10个碳原子且优选2、3或4个碳原子和具有至少1个且优选1至2个双键不饱和位点的直链或支链亚烃基。该术语包括,作为实例,二-乙烯基,烯丙基和丁-3-烯-1-基。该术语中包括顺式和反式异构体或这些异构体的混合物。
“炔基”是指具有2、3、4、5或6个碳原子且优选2至3个碳原子和具有至少1个且优选1至2个三键不饱和位点的直链或支链烃基。这种炔基的实例包括乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。
“亚炔基”是指具有2、3、4、5或6个碳原子且优选2至3个碳原子和具有至少1个且优选1至2个三键不饱和位点的直链或支链亚烃基。该炔基的实例包括乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。
“氨基”是指基团–NH2。
“取代的氨基”是指基团-NRR,其中各R独立地选自氢、烷基、取代的烷基、环烷基、取代的环烷基、烯基、取代的烯基、环烯基、取代的环烯基、炔基、取代的炔基、芳基、杂芳基和杂环基,条件是至少一个R不为氢。
“芳基”是指6、7、8、9、10、11、12、13、14、15、16、17或18个碳原子的一价芳族碳环基团,其具有单环(如以苯基存在)或有多个稠合的环的环体系(该芳环体系的实例包括萘基、蒽基和茚满基),该稠合的环可为或可不为芳族的,条件是连接点是经由芳环的原子。该术语包括,作为实例,苯基和萘基。除非另外被芳基取代基的定义所限制,该芳基可任选取代有1、2、3、4或5个取代基,或1、2或3个取代基,所述取代基选自酰基氧基、羟基、巯基、酰基、烷基、烷氧基、烯基、炔基、环烷基、环烯基、取代的烷基、取代的烷氧基、取代的烯基、取代的炔基、取代的环烷基、取代的环烯基、氨基、取代的氨基、氨基酰基、酰基氨基、烷芳基、芳基、芳基氧基、叠氮基、羧基、羧基酯、氰基、卤素、硝基、杂芳基、杂芳基氧基、杂环基、杂环氧基、氨基酰基氧基、氧基酰基氨基、硫代烷氧基(thioalkoxy)、取代的硫代烷氧基、硫代芳基氧基、硫代杂芳基氧基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-杂芳基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基、-SO2-杂芳基和三卤代甲基。
“环烷基”是指3,4,5,6,7,8,9,或10个碳原子的环烷基,其具有单一环或多个环,包括稠合、桥接和螺环体系。合适的环烷基的实例包括,例如,金刚烷基、环丙基、环丁基、环戊基、环辛基等。该环烷基包括,作为实例,单环结构如环丙基、环丁基、环戊基、环辛基、等,或多环结构如金刚烷基,等。
“杂芳基”是指在环中有1、2、3、4、5、6、7、8、9、10、11、12、13、14、或15个碳原子,如1、2、3、4、5、6、7、8、9或10个碳原子和1、2、3、4、5、6、7、8、9或10个选自氧、氮和硫的杂原子的芳族基。该杂芳基可具有单环(如,吡啶基、咪唑基或呋喃基)或在环体系有多个稠合的环(例如像在以下基团中,如,吲嗪基、喹啉基、苯并呋喃、苯并咪唑基或苯并噻吩基),其中环体系中至少一个环为芳族的且环体系中至少一个环为芳族的,条件是连接点是经由芳环的原子。在某些实施方案中,杂芳基的氮和/或硫环原子任选被氧化以提供N-氧化物(N→O)、亚硫酰基或磺酰基部分。该术语包括,作为实例,吡啶基、吡咯基、吲哚基、噻吩基和呋喃基。除非另外被杂芳基取代基的定义所约束,该杂芳基可任选被1、2、3、4或5个取代基,或1、2或3个取代基取代,所述取代基选自酰基氧基、羟基、巯基、酰基、烷基、烷氧基、烯基、炔基、环烷基、环烯基、取代的烷基、取代的烷氧基、取代的烯基、取代的炔基、取代的环烷基、取代的环烯基、氨基、取代的氨基、氨基酰基、酰基氨基、烷芳基、芳基、芳基氧基、叠氮基、羧基、羧基酯、氰基、卤素、硝基、杂芳基、杂芳基氧基、杂环基、杂环氧基、氨基酰基氧基、氧基酰基氨基、硫代烷氧基、取代的硫代烷氧基、硫代芳基氧基、硫代杂芳基氧基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-杂芳基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基和-SO2-杂芳基、以及三卤代甲基。
杂芳基的实例包括,但不限于,吡咯、咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、吲嗪、异吲哚、吲哚、嘌呤、异喹啉、喹啉、酞嗪、萘基吡啶、喹喔啉、喹唑啉、噌啉、蝶啶、咔唑、咔啉、菲啶、吖啶、菲咯啉、异噻唑、吩嗪、异噁唑、吩噁嗪、吩噻嗪、哌啶、哌嗪、邻苯二甲酰亚胺、4,5,6,7-四氢苯并[b]噻吩、噻唑、噻吩、苯并[b]噻吩,等。
“杂环”、“杂环的”、“杂环烷基”或“杂环基”是指饱和或部分不饱和基团,其具有单环或多个缩合的环,包括稠合、桥接或螺环体系,且具有3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、或20个环原子,包括1、2、3、4、5、6、7、8、9或10个杂原子。这些环原子选自碳、氮、硫或氧,其中,在稠合的环体系中,所述环的一个或多个可为环烷基、芳基或杂芳基,条件是连接点是经由非芳环。在某些实施方案中,杂环基的氮和/或硫原子任选被氧化以提供N-氧化物、-S(O)-或–SO2-部分。
杂环的实例包括,但不限于,氮杂环丁烷、二氢吲哚、吲唑、喹嗪、咪唑烷、咪唑啉、哌啶、哌嗪、二氢吲哚、1,2,3,4-四氢异喹啉、噻唑烷、吗啉基、硫吗啉基(也称为硫代吗啉基(thiamorpholinyl))、1,1-二氧代硫吗啉基、哌啶基、吡咯烷、四氢呋喃基,等。
当杂芳基或杂环基是“取代的”时,除非另外被杂芳基或杂环取代基的定义所约束,这些杂芳基或杂环基可取代有1、2、3、4或5个,或1、2或3个取代基,所述取代基选自烷基、取代的烷基、烷氧基、取代的烷氧基、环烷基、取代的环烷基、环烯基、取代的环烯基、酰基、酰基氨基、酰基氧基、氨基、取代的氨基、氨基酰基、氨基酰基氧基、叠氮基、氰基、卤素、羟基、氧代、硫酮基(thioketo)、羧基、羧基酯、硫代芳基氧基、硫代杂芳基氧基、硫基杂环氧基、巯基、硫代烷氧基、取代的硫代烷氧基、芳基、芳基氧基、杂芳基、杂芳基氧基、杂环基、杂环氧基、羟基氨基、烷氧基氨基、硝基、磺酰基氨基、-SO-烷基、-SO-取代的烷基、-SO-芳基、-SO-杂芳基、-SO-杂环基、-SO2-烷基、-SO2-取代的烷基、-SO2-芳基、-SO2-杂芳基、和-SO2-杂环基。
“聚亚烷基二醇”是指直链或支链的聚亚烷基二醇聚合物如聚乙二醇、聚丙二醇和聚丁二醇。聚亚烷基二醇亚单元为单一聚亚烷基二醇单元。例如,聚乙二醇亚单元的实例为乙二醇-O-CH2-CH2-O-,或丙二醇-O-CH2-CH2-CH2-O-,在链端点以氢封端。其它聚(亚烷基二醇)的实例包括,但不限于,PEG、PEG衍生物如甲氧基聚(乙二醇)(mPEG)、聚(环氧乙烷)、PPG、聚(四亚甲基二醇)、聚(环氧乙烷-共聚-环氧丙烷),或其共聚物和组合。
“聚胺”是指在单体单元中具有胺官能团的聚合物,其或者掺入主链,如聚亚烷基亚胺中那样,或掺入侧基,如聚乙烯基胺那样。
除了本文的公开内容,当用于修饰指定基或基团时,术语“取代的”还可以表示指定基或基团的一个或多个氢原子各自彼此独立地被如下定义的相同的或不同取代基代替。
除了本文关于单独术语公开的基团,在指定基或基团中取代饱和碳原子上一个或多个氢(单个碳上的任何两个氢可被=O、=NR70、=N-OR70、=N2或=S代替)的取代基,除非另有所述,为-R60、卤素、=O、-OR70、-SR70、-NR80R80、三卤代甲基、-CN、-OCN、-SCN、-NO、-NO2、=N2、-N3、-S(O)R70、-SO2R70、-SO2O–M+、-SO2OR70、-OSO2R70、-OSO2O–M+、-OSO2OR70、-P(O)(O–)2(M+)2、-P(O)(OR70)O–M+、-P(O)(OR70)2、-C(O)R70、-C(S)R70、-C(NR70)R70、-C(O)O–M+、-C(O)OR70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OC(O)O-M+、-OC(O)OR70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70CO2–M+、-NR70CO2R70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60选自任选取代的烷基、环烷基、杂环烷基、杂环烷基烷基、环烷基烷基、芳基、芳基烷基、杂芳基和杂芳基烷基,各R70独立地为氢或R60;各R80独立地为R70,或者,两个R80',与它们连接的氮原子一起形成3-、4-、5-、6-、或7-元杂环烷基,该杂环烷基可任选包含1至4个相同或不同的选自O、N和S的其它杂原子,其中N可具有–H、C1-C4烷基、-C(O)C1-4烷基、-CO2C1-4烷基、或–SO2C1-4烷基取代;且各M+为具有净单一正电荷的抗衡离子。各M+可独立地为,例如,碱金属离子,如K+、Na+、Li+;铵离子,如+N(R60)4;或碱土金属离子,如[Ca2+]0.5、[Mg2+]0.5或[Ba2+]0.5("下标0.5是指该二价碱土金属离子的一个抗衡离子可为该实施方案的化合物的离子化形式,而另一典型抗衡离子如氯离子,或者本文公开的两个离子化化合物可以用作这样的二价碱土金属离子的抗衡离子,或该实施方案的双离子化化合物可用作这种二价碱土离子的抗衡离子)。
除了本文的公开内容,在"取代的"烯烃、炔烃、芳基和杂芳基中不饱和碳原子上氢的取代基,除非另有所述,为-R60、卤素、-O-M+、-OR70、-SR70、-S–M+、-NR80R80、三卤代甲基、-CF3、-CN、-OCN、-SCN、-NO、-NO2、-N3、-S(O)R70、-SO2R70、-SO3–M+、-SO3R70、-OSO2R70、-OSO3–M+、-OSO3R70、-PO3-2(M+)2、-P(O)(OR70)O–M+、-P(O)(OR70)2、-C(O)R70、-C(S)R70、-C(NR70)R70、-CO2–M+、-CO2R70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OCO2–M+、-OCO2R70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70CO2–M+、-NR70CO2R70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60、R70、R80和M+如之前所定义,条件是在取代的烯烃或炔烃的情况下,所述取代基不为-O-M+、-OR70、-SR70或-S–M+。
除了本文关于单独术语公开的取代基,在“取代的”杂环烷基和环烷基中氮原子上的氢的取代基,除非另有所述,为-R60、-O-M+、-OR70、-SR70、-S-M+、-NR80R80、三卤代甲基、-CF3、-CN、-NO、-NO2、-S(O)R70、-S(O)2R70、-S(O)2O-M+、-S(O)2OR70、-OS(O)2R70、-OS(O)2O-M+、-OS(O)2OR70、-P(O)(O-)2(M+)2、-P(O)(OR70)O-M+、-P(O)(OR70)(OR70)、-C(O)R70、-C(S)R70、-C(NR70)R70、-C(O)OR70、-C(S)OR70、-C(O)NR80R80、-C(NR70)NR80R80、-OC(O)R70、-OC(S)R70、-OC(O)OR70、-OC(S)OR70、-NR70C(O)R70、-NR70C(S)R70、-NR70C(O)OR70、-NR70C(S)OR70、-NR70C(O)NR80R80、-NR70C(NR70)R70和-NR70C(NR70)NR80R80,其中R60、R70,R80和M+如之前所定义。
除了本文公开的内容,在某一实施方案中,经取代的基团具有1、2、3或4个取代基,1、2或3个取代基,1或2个取代基,或1个取代基。
应理解,在上文所定义的所有取代基团中,本文不欲将通过限定本身具有其他取代基的取代基而得到的聚合物(例如,具有取代的芳基作为取代基的取代的芳基,该取代基本身由取代的芳基取代,该取代的芳基进一步由取代的芳基取代,等)包括在内。在此类情况下,这些取代的最大数目为3。例如,本文特定涵盖的取代的芳基的连续取代限于取代的芳基-(取代的芳基)-取代的芳基。
除非另有说明,否则本文未明确定义的取代基的命名法是通过命名官能团的末端部分然后命名朝向连接点的相邻官能团而获得。例如,取代基“芳烷氧羰基”是指基团(芳基)-(烷基)-O-C(O)-。
关于本文公开的含有一或多个取代基的任何基团,应理解,显然,这些基团不含有在空间上不能实现和/或合成上不可行的任何取代或取代方式。另外,主题化合物包括由这些化合物的取代产生的所有立体化学异构体。
术语“药物可接受的盐”意指对于给药至患者(例如哺乳动物)可接受的盐(对给定剂量方案具有可接受的哺乳动物安全性的含抗衡离子的盐)。此类盐可以来源于药物可接受的无机或有机碱,以及来源于药物可接受的无机或有机酸。“药物可接受的盐”是指来源于本领域中熟知的各种有机及无机抗衡离子的化合物的药物可接受的盐,且仅举例而言,包括钠盐、钾盐、钙盐、镁盐、铵盐、四烷基铵盐等;且当分子含有碱性官能团时,包括有机或无机酸的盐,如盐酸盐、氢溴酸盐、甲酸盐、酒石酸盐、苯磺酸盐、甲磺酸盐、乙酸盐、马来酸盐、草酸盐等。
基团结构图中的波浪线代表基团至母体结构的连接点。
术语“其盐”意指当酸的质子由阳离子(例如金属阳离子或有机阳离子等)替换时所形成的化合物。在适用情况下,该盐为药学上可接受的盐,但此条件对不预期给药至患者的中间体化合物的盐而言并非必需。举例而言,本化合物的盐包括其中通过无机或有机酸来质子化以形成阳离子的那些化合物,其中无机或有机酸的共轭碱作为盐的阴离子组分。
“溶剂合物”是指通过溶剂分子与溶质的分子或离子的组合形成的复合物。溶剂可以是有机化合物,无机化合物或两者的混合物。溶剂的一些实例包括但不限于甲醇,N,N-二甲基甲酰胺,四氢呋喃,二甲基亚砜和水。当溶剂是水时,形成的溶剂合物是水合物。
“立体异构体”是指具有相同原子连接性但在空间中具有不同原子排列的化合物。立体异构体包括顺式-反式异构体、E和Z异构体、对映异构体和非对映异构体。
“互变异构体”是指仅在原子的电子连接和/或在质子位置方面不同的分子的替代形式,例如烯醇-酮和亚胺-烯胺互变异构体,或含有-N=C(H)-NH-环原子排列的杂芳基的互变异构形式,例如吡唑、咪唑、苯并咪唑、三唑和四唑。本领域普通技术人员将认识到其它互变异构的环原子排列是可能的。
应当理解,术语“或其盐或溶剂合物或立体异构体”旨在包括盐,溶剂合物和立体异构体的所有排列,例如主题化合物的立体异构体的药学上可接受的盐的溶剂合物。
如本文所用,药物、化合物、缀合物、药物缀合物、抗体药物缀合物或药物组合物的“有效剂量”或“有效量”是足以实现有益或所需结果的量。对于预防性使用,有益或期望的结果包括诸如消除或降低疾病的风险,减轻其严重性或延迟疾病发作的结果,包括在疾病的发展过程中疾病的生化、组织学和/或行为症状,其并发症和中间病理表型。对于治疗用途,有益或期望的结果包括临床结果,例如减少由疾病导致的一种或多种症状,增加患有疾病的那些人的生活质量,减少治疗疾病所需的其它药物的剂量,例如通过靶向增强另一种药物的效果、延迟疾病的进展,和/或延长存活。在癌症或肿瘤的情况下,有效量的药物可以具有以下效果:减少癌细胞数目;减少肿瘤大小;抑制(即,在一定程度上减缓并且优选停止)癌细胞浸润到外周器官中;抑制(即,在一定程度上减缓并且优选停止)肿瘤转移;在一定程度上抑制肿瘤生长;和/或在一定程度上缓解与疾病相关的一种或多种症状。有效剂量可以一次或多次施用来施用。出于本公开的目的,药物、化合物或药物组合物的有效剂量是足以直接或间接实现预防性或治疗性治疗的量。如在临床背景中所理解的,药物、化合物或药物组合物的有效剂量可以或可以不与另一种药物、化合物或药物组合物联合实现。因此,在给予一种或多种治疗剂的上下文中可以考虑“有效剂量”,并且如果与一种或多种其他药剂联合,可以认为单一试剂以有效量给予,期望的结果可以被实现或被实现。
如本文所用,“给药”是指用于分配治疗剂以治疗病症的通用术语。在一些实施方案中,本公开的药物组合物含有适于使化合物或混合物作为片剂、胶囊或丸剂口服给药的药学上可接受的载体或赋形剂,或使得化合物适于肠胃外、静脉内、皮内、肌内、腹膜内、鼻内、舌下、气管内、吸入、眼、阴道、直肠、皮下或经皮施用的药学上可接受的载体或赋形剂。
有多种给药途径。所选择的具体模式当然取决于所选择的具体药剂、所治疗的具体病症和治疗功效所需的剂量。下面讨论几种给药方式。
本发明的化合物或药物组合物的给药可通过本领域技术人员已知的任何方式实现。给药途径包括但不限于口服、肠胃外、静脉内、肌内、腹腔内、鼻内、舌下、气管内、吸入、皮下、眼、阴道和直肠的。全身途径包括口服和肠胃外。
对于口服给药,可以通过将活性化合物与本领域熟知的药学上可接受的载体组合来容易地配制。这样的载体使得本公开的化合物能够配制为用于由待治疗的受试者口服摄取的片剂、丸剂、糖锭剂、胶囊、液体、凝胶、糖浆、浆料、悬浮液等。任选地,口服制剂还可以配制在用于中和内部酸性条件的盐水或缓冲液中,或者可以在没有任何载体的情况下施用。
当需要将它们全身递送时,化合物可以配制成用于通过注射的肠胃外给药,例如通过推注(bolus injection)或连续输注。用于注射的制剂可以以单位剂型存在,例如在安瓿或多剂量容器中,加入防腐剂。组合物可以采取在油性或水性载体中的悬浮液、溶液或乳液的形式,并且可以含有配制剂如悬浮剂、稳定剂和/或分散剂。
用于肠胃外给药的药物制剂包括水溶性形式的活性化合物的水溶液。此外,活性化合物的悬浮液可以制备为适当的油性注射悬浮液。合适的亲脂性溶剂或载体包括脂肪油如芝麻油,或合成脂肪酸酯,如油酸乙酯或甘油三酯,或脂质体。水性注射悬浮液可以含有增加悬浮液粘度的物质,例如羧甲基纤维素钠、山梨醇或葡聚糖。任选地,悬浮液还可以含有合适的稳定剂或增加化合物的溶解性以允许制备高度浓缩溶液的试剂。或者,活性化合物可以是粉末形式,在使用前用合适的载体例如无菌无热原水重构。
化合物还可以配制成直肠或阴道组合物,例如栓剂或保留灌肠剂,例如含有常规栓剂基质如可可脂或其它甘油酯。
如本文所使用的,“与……联合”是指除了一种治疗方式之外施用另一种治疗方式。因此,“与……联合”是指在向个体施用其它治疗方式之前、期间或之后施用一种治疗方式。
如本文所用,“治疗”是用于获得有益或期望的结果的方法,包括并优选临床结果。为了本公开的目的,有益或期望的临床结果包括但不限于以下的一种或多种:减少(或破坏)癌细胞的增殖,减少由疾病导致的症状,增加患有疾病的人的生活质量,减少治疗疾病所需的其它药物的剂量,延迟疾病的进展和/或延长个体的存活。
如本文所用,“延迟疾病的发展”是指延迟、阻碍、减缓、延迟、稳定和/或推迟疾病(例如癌症)的发展。这种延迟可以具有变化的时间长度,这取决于疾病和/或被治疗的个体的病史。对于本领域技术人员明显的是,足够的或显著的延迟实际上可包括预防,即个体不发展疾病。例如,晚期癌症,例如转移的发展可能被延迟。
“个体”或“受试者”是哺乳动物,更优选人。哺乳动物还包括但不限于农场动物、运动动物、宠物(例如猫、狗、马)、灵长类动物、小鼠和大鼠。“在有需要的个体中治疗癌症”是被鉴定为患有癌症的个体,即个体已经由医生(例如使用本领域众所周知的方法)诊断为患有癌症。在一些实施方案中,需要治疗的个体是怀疑患有或发展癌症的个体。怀疑患有或发展癌症的个体的实例包括但不限于被鉴定为具有与癌症或癌症发展相关的突变的受试者,具有癌症家族史的受试者以及先前或曾经已经治愈癌症的受试者(包括缓解期的癌症患者)。
如本文所用,术语“特异性识别”或“特异性结合”是指可测量的和可再现的相互作用,例如靶标和抗体(或分子或部分)之间的吸引或结合,其在存在包括生物分子在内的分子的异质群体的情况下决定靶标的存在。例如,特异性或优先结合表位的抗体是以比其结合靶标或非靶标表位的其它表位更大的亲和力、活动性、更容易和/或更长的持续时间结合该表位的抗体。还应理解,例如,特异性或优先结合第一靶标的抗体(或部分或表位)可以或可以不特异性或优先结合第二靶标。因此,“特异性结合”或“优先结合”不一定需要(尽管其可以包括)排他性结合。特异性结合靶标的抗体可具有至少约10 3M-1或10 4M-1,有时约10 5M-1或10 6M-1,在其它情况下约10 6M-1或10 7M-1,约10 8M-1至10 9M-1,或约10 10M-1至10 11M-1或更高的缔合常数。多种免疫测定形式可用于选择与特定蛋白质特异性免疫反应的抗体。例如,固相ELISA免疫测定常规用于选择与蛋白质特异性免疫反应的单克隆抗体。关于可用于测定特异性免疫反应性的免疫测试形式和条件的描述,参见例如Harlow和Lane(1988)Antibodies,A Laboratory Manual,Cold Spring Harbor Publications,New York。
如本文所用,术语“癌症”、“肿瘤”、“癌性”和“恶性”是指或描述哺乳动物中通常特征在于不受调节的细胞生长的生理状况。癌症的实例包括但不限于癌,包括腺癌、淋巴瘤、母细胞瘤、黑素瘤和肉瘤。这种癌症的更具体的实例包括鳞状上皮细胞癌、小细胞肺癌、非小细胞肺癌、肺腺癌、肺鳞状细胞癌、胃肠癌、霍奇金淋巴瘤和非霍奇金淋巴瘤、胰腺癌、成胶质细胞瘤、子宫颈癌、神经胶质瘤、卵巢癌、肝癌(liver cancer)如肝脏癌瘤(hepatic carcinoma)和肝癌(hepatoma)、膀胱癌、乳腺癌、结肠癌、结肠直肠癌、子宫内膜或子宫癌、唾液腺癌、肾癌如肾细胞癌和维尔姆斯肿瘤、基底细胞癌、黑素瘤、间皮瘤、前列腺癌、甲状腺癌、睾丸癌、食管癌、胆囊癌和各种类型的头颈癌。
如本文和所附权利要求中所使用的,单数形式“一”、“一个”和“该”包括复数指代,除非上下文另有明确指示例如,提及“抗体”是指一至多个抗体,例如摩尔量,并且包括本领域技术人员已知的等同物,等等。
本文中提及“约”一个值或参数包括(并描述)涉及该值或参数本身的实施方案。例如,涉及“约X”的描述包括对“X”的描述。
应当理解,本文所述的本公开的方面和变化包括“由……组成”和/或“基本上由……组成”的方面和变化。
除非另有定义,本文使用的所有技术和科学术语具有与本公开所属领域的普通技术人员通常理解的相同的含义。尽管与本文所述的那些类似或等同的任何方法和材料也可以用于本公开的实践或测试,现在描述优选的方法和材料。本文提及的所有出版物通过引用并入本文,以公开和描述与所引用的出版物相关的方法和/或材料。
除非另有说明,本实施方案的方法和技术通常根据本领域公知的常规方法进行,并且在本说明书通篇引用和讨论的各种一般和更具体的参考文献中描述。参见例如Loudon,Organic Chemistry,第4版,New York:Oxford University Press,2002,第360-361、1084-1085页;Smith和March,March's Advanced Organic Chemistry:Reactions,Mechanisms,and Structure,5th edition,Wiley-Interscience,2001。
本文使用的命名主题化合物的命名法在本文的实施例中说明。该命名通常使用可商购的AutoNom软件(MDL,San Leandro,Calif.)得到。
应当理解,为了清楚起见,在单独实施方案的背景中描述的本公开的某些特征也可以在单个实施方案中组合提供。相反,为了简洁,在单个实施方案的背景中描述的本公开的各种特征也可以单独地或以任何合适的亚组合提供。涉及由变量表示的化学基团的实施方案的所有组合特别地包括在本公开内容中并且在本文中公开,就好像各个和每个组合单独和明确地公开,只要这样的组合包括的化合物为稳定化合物(即,可以分离、表征和测试生物活性的化合物)。此外,描述这些变量的实施方案中列出的化学基团的所有亚组合也特别包括在本公开中,并且在本文中公开,就如同化学基团的各个和每个这样的亚组合在本文中单独和明确地公开。
本发明提供式(I)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
X为亲水性自降解连接基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键或自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
本发明还提供式(II)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
在一些实施方案中,该靶向部分具有一个或多个用于连接至药物部分的连接位点。例如,靶向部分T可具有多个位点以用于连接至连接基-药物部分(例如,A-L4-L3-L2-X-L1-D)。因此,还提供了式(Ia)的化合物:
或其盐或溶剂合物或立体异构体,其中X,L1,L2,L3,L4和A如式(I)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
本发明还提供式(IIa)的化合物:
或其盐或溶剂合物或立体异构体,其中R1,X,L1,L2,L3,L4和A如式(II)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。
肽连接基
在一些实施方案中,L3为肽连接基。在一些实施方案中,L3为1、2、3、4、5、6、7、8、9或10个氨基酸残基的肽连接基。在某些实施方案中,L3为2、3或4个氨基酸残基的肽连接基。在一些情况下,L3为二肽连接基。
氨基酸残基可为天然存在的或非天然氨基酸残基。术语“天然氨基酸”和“天然存在的氨基酸”是指Ala、Asp、Cys、Glu、Phe、Gly、His、Ile、Lys、Leu、Met、Asn、Pro、Gln、Arg、Ser、Thr、Val、Trp和Tyr。“非天然氨基酸”(即,氨基酸不是天然存在)包括,作为非限制性的实例,高丝氨酸、高精氨酸、瓜氨酸、苯基甘氨酸、牛磺酸、碘化酪氨酸(iodotyrosine)、硒代半胱氨酸、正亮氨酸(“Nle”)、正缬氨酸(“Nva”)、β-丙氨酸、L-或D-萘基丙氨酸、鸟氨酸(“Orn”),等。
氨基酸还包括天然和非天然氨基酸的D-形式。“D-”表示具有“D”(右旋)构型的氨基酸,与天然存在(“L-”)氨基酸中的构型相反。在没有指明具体构型的情况下,本领域技术人员将理解氨基酸是L-氨基酸。然而,氨基酸也可以是D-和L-构型的外消旋混合物。天然和非天然氨基酸可商购(Sigma Chemical Co.;Advanced Chemtech)或使用本领域已知的方法合成。可以基于残基的极性,电荷,溶解性,疏水性,亲水性和/或两亲性性质的相似性进行氨基酸替代,只要保留其生物活性即可。
氨基酸残基序列可以被特异性修饰,使得其将通过一种或多种肿瘤相关蛋白酶从所得的肽基衍生物药物-缀合物选择性地酶断裂。
在某些实施方案中,L3为包含至少一个赖氨酸或至少一个精氨酸残基的肽连接基。
在某些实施方案中,L3为包含选自以下的氨基酸残基的肽连接基:赖氨酸、D-赖氨酸、瓜氨酸、精氨酸、脯氨酸、组氨酸、鸟氨酸和谷氨酰胺。
在某些实施方案中,L3为包含选自以下的氨基酸残基的肽连接基:缬氨酸、异亮氨酸、苯丙氨酸、蛋氨酸、天冬酰胺、脯氨酸、丙氨酸、亮氨酸、色氨酸和酪氨酸。
在某些实施方案中,L3为选自以下的二肽连接基:缬氨酸-瓜氨酸、脯氨酸-赖氨酸、蛋氨酸-D-赖氨酸、天冬酰胺-D-赖氨酸、异亮氨酸-脯氨酸、苯丙氨酸-赖氨酸和缬氨酸-赖氨酸。在某些实施方案中,L3为缬氨酸-瓜氨酸。
在其对特定肿瘤相关蛋白酶的酶断裂的选择性方面,可以设计和优化适于在本公开中使用的许多特异性肽连接基分子。用于本公开的某些肽连接基是针对蛋白酶、组织蛋白酶B和D优化的那些。
亲水性自降解连接基
在本文所述的化合物的一些实施方案中,X为亲水性自降解连接基。
本公开的化合物采用亲水性自降解间隔基部分,其将两个官能部分间隔和共价连接在一起,并引入亲水基团,其提供化合物的更好的溶解性。在一些实施方案中,亲水性自降解间隔基部分将靶向部分和药物部分连接在一起。一些酶不稳定连接基的增加的相关疏水性可导致药物缀合物的聚集,特别是对于强疏水性药物。通过将亲水基团引入连接基中,药物缀合物的聚集将减少。
自降解间隔基可以定义为双官能化学部分,其能够将两个间隔开的化学部分共价连接在一起成为通常稳定的三部分分子,可以通过酶断裂从三部分分子中释放一个间隔的化学部分;并且在酶断裂后,可以自发地从分子的其余部分断裂以释放另一个间隔的化学部分。
在某些实施方案中,X为苄基氧基羰基。在某些实施方案中,X为
其中R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基。
在一些实施方案中,本发明提供式(II)的化合物:
或其盐或溶剂合物或立体异构体;
其中:
D为药物部分;
T为靶向部分;
R1为氢、未取代或取代的C1-3烷基、或未取代或取代的杂环基;
L1为键、自降解连接基或环化自消除型连接基;
L2为键、自降解连接基;
其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;
其中如果L2为自降解连接基,则L1为键;
L3为肽连接基;
L4为键或间隔基;且
A为酰基单元。
还提供了式(IIa)的化合物:
或其盐或溶剂合物或立体异构体;其中D、T、L1、L2、L3、L4和A如式(II)所定义,且p为1至20。在一些实施方案中,p为1至8。在一些实施方案中,p为1至6。在一些实施方案中,p为1至4。在一些实施方案中,p为2至4。在一些实施方案中,p为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。在一些实施方案中,p为1、2、3或4。在一些实施方案中,p为2。在一些实施方案中,p为3。在一些实施方案中,p为4。
在式(II)或(IIa)的某些实施方案中,R1为氢。在一些情况下,R1为甲基。
药物部分的释放是基于氨基苄基氧基羰基的自消除反应。出于解释目的,连接有药物和肽的氨基苄基氧基羰基的反应方案示于以下。
方案1
参考方案1,在从肽断裂后,形成氨基苄基氧基羰基且其能经历自发的1,6消除以形成环己-2,5-二烯亚胺衍生物和二氧化碳且释放药物。
任选的第二自降解连接基或环化自消除型连接基
任选的第二自降解连接基或环化自消除型连接基提供另外的连接基以允许精细调整化合物的断裂以释放药物部分。
在本文所述的化合物中,L1为键、自降解连接基或环化自消除型连接基;L2为键或自降解连接基;其中如果L1为自降解连接基或环化自消除型连接基,则L2为键;且其中如果L2为自降解连接基,则L1为键。因此,存在任选的第二自降解连接基或环化自消除型连接基与该亲水性自降解连接基相邻。
在某些实施方案中,L1为键且L2为键。在某些实施方案中,L1为自降解连接基或环化自消除型连接基且L2为键。在某些实施方案中,L1为键且L2为自降解连接基。
在一些实施方案中,L1为键。在某些实施方案中,L1为自降解间隔基或环化自消除型连接基,其将亲水性自降解连接基和药物部分分离。在某些实施方案中,L1为氨基苄基氧基羰基连接基。
在某些实施方案中,L1选自:
其中n为1或2。
在一些情况下,该自降解连接基或环化自消除型连接基为可使用的更多种的部分提供设计可能性。例如,在式(IV)或(IVa)中,亲水性自降解连接基和药物部分之间的氨基甲酸酯连接(-O-C(O)-N(H)-)将提供稳定的药物缀合物且将易于断裂以提供游离药物部分。该亲水性自降解连接基通常以氧基羰基(-O-C(O)-)结尾。如果该药物部分具有可用于反应以形成氨基甲酸酯基的氨基-反应性基团,则该(任选的)第二自降解单元或环化自消除型连接基不是必需的;但其仍可使用。然而,如果该药物不包含氨基,而是包含一些其它反应性官能团,则该药物仍可通过在药物部分和氨基苄基氧基羰基之间包含中间的自降解间隔基或环化自消除型连接基,而掺入本实施方案的包含氨基苄基氧基羰基的化合物中。
以下L1的环化自消除型连接基提供包含羟基的或包含巯基的药物部分与亲水性自降解连接基的氨基苄基氧基羰基的连接:
该实施方案的化合物中的环化自消除型连接基提供化合物的断裂以释放药物部分。相邻亲水性自降解连接基的消除机理将释放L1的氨基。该氨基然后可在环化反应中与L1的氨基甲酸酯基或硫代氨基甲酸酯连接和药物部分反应以释放含羟基或含巯基的药物部分。
在一些实施方案中,L2为键。在某些实施方案中,L2为自降解间隔基,其将亲水性自降解连接基与肽连接基分开。在某些实施方案中,L2为氨基苄基氧基羰基连接基。
在某些实施方案中,L2选自
其中n为1或2。
任选的间隔基
在一些实施方案中,L4为键或间隔基。在某些实施方案中,L4为键。在某些实施方案中,L4为间隔基,其可提供在药物部分和靶向部分之间的距离。
在某些实施方案中,间隔基选自烷基、取代的烷基、烯基、取代的烯基、炔基、取代的炔基、芳基、取代的芳基、环烷基、取代的环烷基、杂芳基、取代的杂芳基、杂环基、取代的杂环基、和杂原子,及其组合。该间隔基在其原子组成中可为同质的或异质的(例如,仅包含碳原子的间隔基或在间隔基上存在碳原子和一种或多种杂原子的间隔基)。优选地,该间隔基包含1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、或50个碳原子和0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30个选自氧、氮和硫的杂原子。该间隔基也可为手性或非手性的,直链、支链的或环状的。
在某些实施方案中,L4为选自以下的间隔基:聚亚烷基二醇、亚烷基、亚烯基、亚炔基和聚胺。亚烯基的实例包括,但不限于,亚乙烯基(-CH=CH-)、亚烯丙基(-CH2C=C-)和亚丁-3-烯-1-基(-CH2CH2C=CH-)。亚烯基的实例包括,但不限于,亚乙炔基(-C≡C-)和亚炔丙基(-CH2C≡C-)。
在某些实施方案中,L4为间隔基,其包含可提供与肽键末端的连接的官能团。官能团,如C(O)、C(O)-NH、S(O)2和S(O)2-NH,可提供与肽键末端的连接。在一些情况下,L4为L4a-C(O)、L4a-C(O)-NH、L4a-S(O)2、L4a-S(O)2-NH,其中L4a选自聚亚烷基二醇、亚烷基、亚烯基、亚炔基和聚胺。在一些情况下,L4为L4a-C(O),其中L4a选自聚亚烷基二醇、亚烷基、亚烯基、亚炔基和聚胺。
在某些实施方案中,L4为L4a-C(O),其中L4a为聚亚烷基二醇。在某些实施方案中,L4为L4a-C(O),其中L4a为聚乙二醇。在某些实施方案中,所述间隔基为式-CH2-(CH2-O-CH2)m-CH2-C(O)-,其中m为整数0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30。
在某些实施方案中,L4为L4a-C(O),其中L4a为亚烷基。在某些实施方案中,L4为L4a-C(O),其中L4a为C1-10亚烷基、C1-8亚烷基或C1-6亚烷基。在某些实施方案中,L4为L4a-C(O),其中L4a为C4亚烷基、C5亚烷基或C6亚烷基。在某些实施方案中,L4为L4a-C(O),其中L4a为C5亚烷基。
酰基单元
在本文所述的化合物中,A为酰基单元。在某些实施方案中,该酰基单元“A”包含硫原子且通过衍生自靶向部分的硫原子连接至靶向部分。在该情况下,二硫键在酰基单元和靶向部分之间形成。
在某些实施方案中,A选自
其中Q2为NH或O,各q独立地为1至10的整数,且各q1独立地为1至10的整数。在一些实施方案中,q为整数2至5,如2、3、4或5。在一些实施方案中,q1为整数2至5,如2、3、4或5。
在某些实施方案中,A为其中Q2为NH或O且q为整数1、2、3、4、5、6、7、8、9或10。在某个情况下,q为2至5的数,如2、3、4或5。
在某些实施方案中,A为其中Q2为NH或O且q为整数1、2、3、4、5、6、7、8、9或10。在某个情况下,q为2至5的数,如2、3、4或5。
在某些实施方案中,A选自
其中Q2为NH或O。
药物部分
本实施方案的药物缀合物对于通常目的是有效的(其中相应药物是有效的),并有卓越的功效,因为其在靶向部分所固有的将药物输送到期望的细胞(在该细胞中,所述药物是特别有益的)的能力。
用于本发明实施方案的优选药物是细胞毒性药物,例如用于癌症治疗的那些。这样的药物通常包括DNA损伤剂、抗代谢物、天然产物及其类似物。某些类型的细胞毒性剂包括例如酶抑制剂,例如二氢叶酸还原酶抑制剂、胸苷酸合酶抑制剂、DNA嵌入剂、DNA断裂剂、拓扑异构酶抑制剂、蒽环类家族药物、长春花药物、丝裂霉素、博莱霉素、细胞毒性核苷、蝶啶家族药物、二炔类、鬼臼毒素、分化诱导剂和紫杉醇。这些类别的某些有用成员包括例如甲氨蝶呤、甲基叶酸、二氯甲氨蝶呤、5-氟尿嘧啶、6-巯基嘌呤、阿糖胞苷、美法仑、长春罗新、白诺西丁(leurosideine)、放线菌素、柔红霉素、多柔比星、丝裂霉素C、丝裂霉素A、洋红霉素、氨基蝶呤、他利霉素、鬼臼毒素和鬼臼毒素衍生物如依托泊苷或磷酸依托泊苷、长春碱、长春新碱、长春地辛、紫杉醇、泰索帝、视黄酸、丁酸、N8-乙酰基亚精胺、喜树碱、以及它们的类似物。其它药物包括多拉司他汀和多卡霉素。
本领域技术人员可对所需化合物进行化学修饰,以使该化合物的反应对于制备本公开的缀合物更为方便。
在某些实施方案中,D为具有化学反应性官能团的药物部分,通过该化学反应性官能团该药物连接至L1或X。在一些情况下,该官能团选自伯胺、仲胺、羟基和巯基。在一些情况下,该官能团为伯胺或仲胺。在一些情况下,该官能团为羟基。在一些情况下,该官能团为巯基。
如上所述,该亲水性自降解连接基通常以氧基羰基(-O-C(O)-)终止。因此,含氨基的药物部分将易于与氧基羰基反应以形成氨基甲酸酯基。在某些实施方案中,D为含氨基的药物部分,其中该药物通过氨基连接至L1或X。
然而,如果该药物部分不包含氨基,则L1的第二自降解连接基或环化自消除型连接基可为更多种可使用的部分提供设计可能性。在某些实施方案中,D为含羟基或含巯基的药物部分,其中该药物通过羟基或巯基连接至L1。
代表性含氨基的药物包括丝裂霉素-C、丝裂霉素-A、柔红霉素、多柔比星、氨基蝶呤、放线菌素、博来霉素、9-氨基喜树碱、N8-乙酰基亚精胺、1-(2-氯乙基)-1,2-二甲烷磺酰基酰肼、他利霉素、阿糖胞苷、多拉司他汀以及它们的衍生物。含氨基的药物还包括天然不包含氨基的药物的氨基衍生物。在某些实施方案中,D为多卡霉素、多拉司他汀、tubulysin、多柔比星(DOX)、紫杉醇或丝裂霉素C(MMC),或其氨基衍生物。
代表性含羟基的药物包括依托泊苷,喜树碱、紫杉醇、埃斯培拉霉素、1,8-二羟基-双环[7.3.1]十三碳-4-9-二烯-2,6-二炔-13-酮(美国专利5,198,560)、鬼臼毒素、anguidine、长春新碱、长春碱、吗啉-多柔比星、正-(5,5-二乙酰氧基-戊基)多柔比星、多卡霉素,和它们的衍生物。
代表性含巯基的药物包括埃斯培拉霉素和6-疏基嘌呤,和它们的衍生物。
用作本实施方案中的药物的一组的细胞毒素剂包括下式的药物:
(多拉司他汀的氨基衍生物)
(多卡霉素的氨基衍生物)
(多卡霉素的氨基衍生物)。
靶向部分
如本公开中所述的靶向部分是指与给定细胞群体特异性结合、复合、反应或缔合的部分或分子。例如,靶向部分可以和与给定细胞群体(例如,寻求治疗性处理或以其它方式生物修饰的给定细胞群体)相关的接受部分或受体特异性结合、复合、反应或缔合。在本文所述的缀合物中,本文所述的靶向部分通过连接基与缀合物中的药物部分连接。在一些实施方案中,靶向部分能够将药物部分(例如,用于治疗目的的药物部分)递送至靶向部分与其结合、复合、反应或缔合的特定靶细胞群。
该靶向部分可包括,例如,大分子量蛋白质,例如,抗体、较小分子量蛋白质、多肽或肽、和非肽基部分。本文所述的蛋白质、多肽或肽部分可包括,例如,转铁蛋白、血清白蛋白、表皮生长因子("EGF")、铃蟾肽(bombesin)、胃泌素、胃泌素-释放肽、血小板-衍生的生长因子、IL-2、IL-6、肿瘤生长因子("TGF")如TGF-α和TGF-β、痘苗病毒生长因子("VGF")、胰岛素和胰岛素样生长因子I和II。非肽基部分可包括,例如,碳水化合物、凝集素和源自低密度脂蛋白的载脂蛋白。在某些实施方案中,蛋白质、抗体、多肽或肽可以指其未修饰的形式、已被修饰以用于本文所述的缀合物中的形式,例如用于与连接基结合的形式,或者在本文所述的缀合物中的部分。
在一些实施方案中,该靶向部分为抗体(或抗体部分或抗体靶向部分)。在一些实施方案中,该靶向部分包括抗体。在一些实施方案中,该靶向部分包括巯基(-SH)基团(例如,游离反应性巯基(-SH)基团)或可被修饰以包含该巯基。在一些实施方案中,该靶向部分包括具有巯基(例如,游离反应性巯基)的抗体。在一些实施方案中,该靶向部分包括游离巯基如具有游离巯基的抗体或可被修饰以包含该巯基。在一些实施方案中,包含巯基或硫醇基的该靶向部分通过巯基中的硫原子连接至连接基。
在一些实施方案中,该靶向部分(例如,抗体靶向部分)具有一个或多个连接位点以用于连接至药物部分。例如,靶向部分T(例如,抗体)可具有多个位点(例如,多个巯基)以用于连接至连接基-药物部分(例如,A-L4-L3-L2-X-L1-D,其中A适合于连接至靶向抗体的巯基)。在一些实施方案中,该靶向部分可具有1至20个连接位点。在一些实施方案中,该靶向部分可具有1至20,1至10,1至8,1至6,1至4,2至8,2至6,或2至4个连接位点。在一些实施方案中,该靶向部分具有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个连接位点。在一些实施方案中,该靶向部分具有1,2,3,4,5,6,7,或8个连接位点。在一些实施方案中,该靶向部分具有2个连接位点。在一些实施方案中,该靶向部分具有1个连接位点。在一些实施方案中,该靶向部分具有4个连接位点。在一些情况下,某些潜在的连接位点可能不适于连接至药物部分。因此,靶向部分T中的连接位点的数目可导致药物缀合物具有比潜在的连接位点的数目更少数量的连接药物部分。在一些实施方案中,一个或多个连接位点可以用于结合药物部分。例如,抗体靶向部分可以在抗体的每条链上具有一个或两个巯基,其适合通过连接基与药物部分结合。
在一些实施方案中,该靶向部分是抗体或抗体靶向部分。本文所述的抗体是指能够通过位于免疫球蛋白分子的可变区中的至少一个抗原识别位点特异性结合靶标(例如碳水化合物、多核苷酸、脂质、多肽等)的免疫球蛋白分子。如本文所用,术语“抗体”不仅包括完整的多克隆或单克隆抗体,而且包括其抗原结合片段(例如Fab、Fab’、F(ab’)2、Fv)、单链(ScFv)、其突变体、包含抗体部分的融合蛋白、以及包含抗原识别位点的免疫球蛋白分子的任何其它修饰的构型。抗体包括任何类别的抗体,例如IgG、IgA或IgM(或其亚类),并且抗体不需要是任何特定类别。根据其重链恒定结构域的抗体氨基酸序列,免疫球蛋白可以归于不同类别。有五种主要类型的免疫球蛋白:IgA、IgD、IgE、IgG和IgM,其中几种可以进一步分为亚类(同种型),例如IgG1、IgG2、IgG3、IgG4、IgA1和IgA2。对应于不同类别的免疫球蛋白的重链恒定结构域分别称为α、δ、ε、γ和μ。不同类别的免疫球蛋白的亚基结构和三维构型是公知的。
包括或用于本文所述的靶向部分(或抗体靶向部分)中的抗体可包括单克隆抗体、多克隆抗体、抗体片段(例如Fab、Fab’、F(ab’)2、Fv、Fc等)、嵌合抗体、人源化抗体、人抗体(例如完全人抗体)、单链(ScFv)、双特异性抗体、多特异性抗体、其突变体、包含抗体部分的融合蛋白、以及包含所需特异性的抗原识别位点的免疫球蛋白分子的任何其他修饰的构型。抗体可以是鼠、大鼠、骆驼、人或任何其它来源(包括人源化抗体)。在一些实施方案中,用于本文所述的靶向部分(或抗体靶向部分)中的抗体是以下任一种:对多肽具有亲和力的双特异性抗体、多特异性、单链、双功能和嵌合和人源化分子,该亲和力由所述抗体的至少一个高变区(HVR)或互补决定区(CDR)赋予。本公开中使用的抗体还包括单结构域抗体,其是抗体重链的可变结构域或抗体轻链的可变结构域。Holt等人,Trends Biotechnol.21:484-490,2003。现有技术也已知制备包含抗体重链的可变结构域或抗体轻链的可变结构域的域抗体的方法,其包含6种天然存在的来自抗体的HVR或CDR中的3种。参见,例如,Muyldermans,Rev.Mol.Biotechnol.74:277-302,2001。
在一些实施方案中,包括或用于本文所述的靶向部分(或抗体靶向部分)的抗体是单克隆抗体。如本文所用,单克隆抗体是指基本上同质性抗体的抗体,即构成群体的单个抗体是相同的,除了可能以少量存在的可能的天然存在的突变。此外,与通常包含针对不同决定簇(表位)的不同抗体的多克隆抗体制剂相反,单克隆抗体不是离散抗体的混合物。修饰语“单克隆”表示抗体从基本上同质的抗体群获得的特征,并且不应解释为需要通过任何特定方法产生抗体。例如,本公开中使用的单克隆抗体可以通过由Kohler和Milstein,1975,Nature,256:495首先描述的杂交瘤方法制备,或者可以通过重组DNA方法制备,如美国专利4,816,567所述。单克隆抗体还可以从例如使用McCafferty等人,1990,Nature,348:552-554中描述的技术产生的噬菌体库中分离。
在一些实施方案中,包括或用于本文所述的靶向部分(或抗体靶向部分)的抗体是嵌合抗体。如本文所用,嵌合抗体指具有来自第一物种的可变区或可变区的部分和来自第二物种的恒定区的抗体。完整的嵌合抗体包含两个拷贝的嵌合轻链和两个拷贝的嵌合重链。嵌合抗体的产生是本领域已知的(Cabilly等人(1984),Proc.Natl.Acad.Sci.USA,81:3273-3277;Harlow和Lane(1988),Antibodies:a Laboratory Manual,Cold Spring Harbor Laboratory)。通常,在这些嵌合抗体中,轻链和重链的可变区都模拟来源于一种哺乳动物的抗体的可变区,而恒定部分与来源于另一种的抗体的序列同源。这种嵌合形式的一个明显的优点是,例如,可变区可以方便地从目前已知的来源使用容易获得的杂交瘤或来自非人宿主生物的B细胞与来源于例如人细胞制备物的恒定区组合而衍生。尽管可变区具有易于制备的优点,并且特异性不受其来源的影响,但是相比来自非人源的恒定区,当注射抗体时,作为人的恒定区不太可能引起来自人类受试者的免疫应答。然而,该定义不限于该特定示例。
在一些实施方案中,包括或用于本文所述的靶向部分(或抗体靶向部分)的抗体是人源化抗体。如本文所用,人源化抗体是指特异性嵌合免疫球蛋白、免疫球蛋白链或其片段(例如Fv、Fab、Fab'、F(ab')2或抗体的其它抗原结合亚序列)的非人(例如鼠)抗体,其含有源自非人免疫球蛋白的最小序列。大多数情况下,人源化抗体是人免疫球蛋白(受体抗体),其中来自受体的HVR或CDR的残基被来自非人物种(供体抗体)的HVR或CDR的残基替换,该非人物种例如具有所需特异性、亲和力和能力的小鼠,大鼠,或兔。在一些情况下,人免疫球蛋白的Fv构架区(FR)残基被相应的非人残基替换。此外,人源化抗体可以包含既不在受体抗体中也不在输入的HVR或CDR或框架序列中发现,但被包括以进一步改进和优化抗体性能的残基。通常,人源化抗体将包含至少一个、通常两个可变结构域的基本上全部,其中所有或基本上所有HVR或CDR区对应于非人免疫球蛋白的那些,并且全部或基本上全部FR区是人免疫球蛋白共有序列的FR区。人源化抗体最佳地还将包含免疫球蛋白恒定区或结构域(Fc)的至少一部分,通常是人免疫球蛋白的恒定区或结构域。抗体可具有如WO 99/58572中所述修饰的Fc区。其他形式的人源化抗体具有相对于原始抗体改变的一个或多个HVR或CDR(一个,两个,三个,四个,五个,六个),其也称为“衍生自”一个或更多个来自原始抗体的HVR或CDR的一个或多个HVR或CDR。
在一些实施方案中,包括或用于本文所述的靶向部分(或抗体靶向部分)的抗体是人抗体。如本文所用,人抗体指具有对应于由人产生的抗体的氨基酸序列的抗体,和/或已使用本领域已知的用于制备人抗体的任何技术制备的抗体。本文使用的人抗体包括包含至少一个人重链多肽或至少一个人轻链多肽的抗体。一个这样的实例是包含鼠轻链和人重链多肽的抗体。人抗体可以使用本领域已知的多种技术产生。在一个实施方案中,人抗体选自噬菌体库,其中噬菌体库表达人抗体(Vaughan等人,1996,Nature Biotechnology,14:309-314;Sheets等人,1998,PNAS,95:6157-6162;Hoogenboom和Winter,1991,J.Mol.Biol.,227:381;Marks等人,1991,J.Mol.Biol.,222:581)。人抗体也可以通过将人免疫球蛋白基因座导入转基因动物(例如其中内源免疫球蛋白基因已部分或完全失活的小鼠)来制备。该方法描述于美国专利5,545,807;5,545,806;5,569,825;5,625,126;5,633,425;和5,661,016。或者,人抗体可以通过使产生针对靶标抗原的抗体的人B淋巴细胞永生化来制备(所述B淋巴细胞可以从个体中回收或可以在体外免疫)。参见,例如,Cole等人,Monoclonal Antibodies and Cancer Therapy,Alan R.Liss,p.77(1985);Boerner等,1991,J.Immunol.,147(1):86-95;和美国专利号5,750,373。
人表皮生长因子2蛋白,HER2(ErbB2)受体酪氨酸激酶,已知在发育和肿瘤发生中都发挥关键作用。在一些实施方案中,包括或用于本文所述的靶向部分(或抗体靶向部分)的抗体特异性结合HER2。在进一步的实施方案中,抗HER2抗体是单克隆或人源化抗体。人源化单克隆抗HER2抗体曲妥珠单抗目前用于治疗HER2-阳性癌症。在一些实施方案中,包括在或用于本文所述的靶向部分中的抗体是曲妥珠单抗。其它抗HER2抗体,包括帕妥珠单抗和margetuximab,也是本领域已知的。在一些实施方案中,包括在或用于本文所述的靶向部分中的抗体是帕妥珠单抗。在一些实施方案中,包括在或用于本文所述的靶向部分中的抗体是margetuximab。
在一些实施方案中,该抗HER2抗体包含轻链可变区和/或重链可变区,该含轻链可变区包含1、2或3个源自SEQ ID NO:7的HVR(或CDR),该重链可变区包含1、2或3个源自SEQ ID NO:8的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含所述3个源自SEQ ID NO:7的HVR(或CDR),该重链可变区包含所述3个源自SEQ ID NO:8的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含的氨基酸序列至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、或至少约99%与SEQ ID NO:7的序列相同,该重链可变区包含的氨基酸序列至少约85%、至少约86%、至少约87%、至少约88%、至少约89%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%、或至少约99%与SEQ ID NO:8的序列相同。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含SEQ ID NO:7的氨基酸序列,该重链可变区包含SEQ ID NO:8的氨基酸序列。
在一些实施方案中,该抗HER2抗体包含轻链可变区和/或重链可变区,该轻链可变区包含1、2或3个源自SEQ ID NO:12的HVR(或CDR),该重链可变区包含1、2或3个源自SEQ ID NO:13的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含所述3个源自SEQ ID NO:12的HVR(或CDR),该重链可变区包含所述3个源自SEQ ID NO:13的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含的氨基酸序列至少约85%,至少约86%,至少约87%,至少约88%,至少约89%,至少约90%,至少约91%,至少约92%,至少约93%,至少约94%,至少约95%,至少约96%,至少约97%,至少约98%,或至少约99%与SEQ ID NO:12的序列相同,该重链可变区包含的氨基酸序列至少约85%,至少约86%,至少约87%,至少约88%,至少约89%,至少约90%,至少约91%,至少约92%,至少约93%,至少约94%,至少约95%,至少约96%,至少约97%,至少约98%,或至少约99%与SEQ ID NO:13的序列相同。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含SEQ ID NO:12的氨基酸序列,该重链可变区包含SEQ ID NO:13的氨基酸序列。
在一些实施方案中,该抗HER2抗体包含轻链可变区和/或重链可变区,该轻链可变区包含1、2或3个源自SEQ ID NO:14的HVR(或CDR),该重链可变区包含1、2或3个源自SEQ ID NO:15的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含所述3个源自SEQ ID NO:14的HVR(或CDR),该重链可变区包含所述3个源自SEQ ID NO:15的HVR(或CDR)。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含的氨基酸序列至少约85%,至少约86%,至少约87%,至少约88%,至少约89%,至少约90%,至少约91%,至少约92%,至少约93%,至少约94%,至少约95%,至少约96%,至少约97%,至少约98%,或至少约99%与SEQ ID NO:14的序列相同,该重链可变区包含的氨基酸序列至少约85%,至少约86%,至少约87%,至少约88%,至少约89%,至少约90%,至少约91%,至少约92%,至少约93%,至少约94%,至少约95%,至少约96%,至少约97%,至少约98%,或至少约99%与SEQ ID NO:15的序列相同。在一些实施方案中,该抗体包含轻链可变区和/或重链可变区,该轻链可变区包含SEQ ID NO:14的氨基酸序列,该重链可变区包含SEQ ID NO:15的氨基酸序列。
表1:抗HER2ADC的序列ID NO.
人κ轻链恒定域序列(SEQ ID NO.1)RTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
人IgG1重链恒定域序列(SEQ ID NO.2)
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
人IgG2重链恒定域序列(SEQ ID NO.3)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPSK
人IgG3重链恒定域序列(SEQ ID NO.4)
ASTKGPSVFPLAPCSRSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYTCNVNHKPSNTKVDKRVELKTPLGDTTHTCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPEPKSCDTPPPCPRCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFKWYVDGVEVHNAKTKPREEQYNSTFRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESSGQPENNYNTTPPMLDSDGSFFLYSKLTVDKSRWQQGNIFSCSVMHEALHNRFTQKSLSLSPGK
人IgG4重链恒定域序列(SEQ ID NO.5)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPSCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
hIgG4-S228P的氨基酸序列(SEQ ID NO.6)
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
曲妥珠单抗(Roche Inc.)轻链可变区的氨基酸序列(SEQ ID NO.7)
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIK
曲妥珠单抗(Roche Inc.)重链可变区的氨基酸序列(SEQ ID NO.8)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSS
包含人κ恒定域的曲妥珠单抗(Roche Inc.)轻链的氨基酸序列(SEQ ID NO.9)
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
包含人IgG1恒定域的曲妥珠单抗(Roche Inc.)重链的氨基酸序列(SEQ ID NO.10)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
包含人IgG4-S228P恒定域的曲妥珠单抗(Roche Inc.)重链的氨基酸序列(SEQ ID NO.11)
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGK
帕妥珠单抗(Roche Inc.)轻链可变区的氨基酸序列(SEQ ID NO.12)
DIQMTQSPSSLSASVGDRVTITCKASQDVSIGVAWYQQKPGKAPKLLIYSASYRYTGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYIYPYTFGQGTKVEIK
帕妥珠单抗(Roche Inc.)重链可变区的氨基酸序列(SEQ ID NO.13)
EVQLVESGGGLVQPGGSLRLSCAASGFTFTDYTMDWVRQAPGKGLEWVADVNPNSGGSIYNQRFKGRFTLSVDRSKNTLYLQMNSLRAEDTAVYYCARNLGPSFYFDYWGQGTLVTVSS
Margetuximab(Macrogenics Inc.)轻链可变区的氨基酸序列(SEQ ID NO.14)
DIVMTQSHKFMSTSVGDRVSITCKASQDVNTAVAWYQQKPGHSPKLLIYSASFRYTGVPDRFTGSRSGTDFTFTISSVQAEDLAVYYCQQHYTTPPTFGGGTKVEIK
Margetuximab(Macrogenics Inc.)重链可变区的氨基酸序列(SEQ ID NO.15)
QVQLQQSGPELVKPGASLKLSCTASGFNIKDTYIHWVKQRPEQGLEWIGRIYPTNGYTRYDPKFQDKATITADTSSNTAYLQVSRLTSEDTAVYYCSRWGGDGFYAMDYWGQGASVTVSS
表2:抗HER2抗体曲妥珠单抗、帕妥珠单抗和Margetuximab的CDR的氨基酸序列
在一些实施方案中,包括在或用于本文所述的靶向部分中的抗体被修饰。在一些实施方案中,所述修饰为工程化的半胱氨酸取代。
在一些实施方案中,工程化的半胱氨酸取代发生在抗体的IgG重链上。在一些实施方案中,工程化的半胱氨酸取代发生在抗体的IgG重链上的特定位置。在一些实施方案中,可以具有工程化的半胱氨酸取代的IgG重链上的氨基酸位置包括(EU编号)118-215、234、235、236、237、238、239、246、248、249、254、265、267、269、270、273、276、278、279、282、283、284、286、287、289、292、293、294、297、298、299、300、302、303、312、314、315、318、320、324、326、327、330、332、333、334、335、336、337、339、341-447。上述公开的氨基酸位置描述于US 2012/0148580 A1;WO 2013/093809 A1;US 2009/0258420 A1;US 7521541 B2;US 7855275 B2;US 2011/0137017 A1;US 2012/0213705 A1;US 2011/0033378 A1;US 8455622 B2,其全部内容通过引用并入本文。可以是用于位点特异性缀合的工程化的半胱氨酸的IgG重链上的其它位置包括(EU编号)121、122、124、125、126、129、159、187、188、190、191、193、197、199、201、202、203、205、207、208、209、211、212、215、295、296、301。
在一些实施方案中,工程化的半胱氨酸取代发生在抗体的IgG轻链上。在一些实施方案中,工程化的半胱氨酸取代发生在抗体的IgG轻链上的特定位置。在一些实施方案中,可具有工程化的半胱氨酸取代的IgG轻链上的氨基酸位置包括(Kabat编号)108-211,如WO 2013/093809 A1;US 2009/0258420 A1;US 7855275B2;US 8455622B2中所述,其全部内容通过引用并入本文。可以是用于位点特异性缀合的工程化的半胱氨酸的IgG轻链上的另外的位置包括(Kabat编号)112、114、115、116、147、195、199、200、201、202、203、206、207、208、209、210。
治疗应用
在一些实施方案中,本公开提供了通过向细胞施用足够致死量的本文所讨论的化合物来杀死细胞的方法。在一些实施方案中,被杀死的细胞是癌细胞。在一些实施方案中,所述细胞是乳腺癌细胞,或胃癌细胞或卵巢癌细胞。在一些实施方案中,本发明公开的杀死细胞的方法可以在体外进行。在一些实施方案中,杀死细胞的方法可以在体内进行。
在一些实施方案中,本文所讨论的化合物可以作为治疗受试者中疾病或病症的治疗方案的一部分以有效剂量施用。在一些实施方案中,本公开提供了用于治疗有需要的个体中的癌症的方法,其包括向个体施用有效剂量的本文所公开的化合物。在一些实施方案中,有效剂量为约0.001mg/kg至约1000mg/kg,约0.01mg/kg至约750mg/kg,约0.1mg/kg至约500mg/kg,约1.0mg/kg至约250mg/kg,约10.0mg/kg至约150mg/kg,一次或多次给药,持续一天或几天或多天,这取决于给药方式和上述因素。
在一些实施方案中,本公开的化合物可以与其它治疗化合物组合施用。在一些实施方案中,其它治疗化合物是抗癌药物或化学治疗剂。本领域普通技术人员将熟悉多种癌症化疗剂。在一些实施方案中,本公开的化合物可以与其它形式的癌症治疗例如(但不限于)放射治疗组合施用。
在一些实施方案中,本公开的治疗癌症的方法的使用导致有益的或期望的临床结果,包括但不限于减少(或破坏)癌细胞的增殖,减少由疾病导致的症状,增加那些患有疾病的人的生活质量,和/或延缓疾病的发展。对于本领域技术人员明显的是,足够的或显著的延迟实际上可包括预防,即该个体不产生癌症。例如,晚期癌症,例如转移的发展可能被延迟。
在一些实施方案中,本文所述的化合物的靶向部分特异性结合癌细胞。癌症结合靶向部分的示例性但非限制性实例包括抗CD20抗体,抗CD30抗体和抗HER2抗体。在进一步的实施方案中,本文所讨论的化合物的药物部分是有效治疗癌症的药物。这种药物的非限制性实例包括丝裂霉素-C、丝裂霉素-A、柔红霉素、多柔比星、氨基蝶呤、放线菌素、博来霉素、9-氨基喜树碱、N8-乙酰基亚精胺、1-(2-氯乙基)-1,2-二甲烷磺酰基酰肼、他利霉素、阿糖胞苷、多拉司他汀以及它们的衍生物。
提供以下实施例以说明但不限制本公开的效用。
实施例1
实施例2的材料和方法
连接基-药物的合成
化合物Tap-18H的合成示于以下方案。中间体化合物M和O的合成也示于以下方案。
化合物TAP-18H的合成
化合物M的合成
化合物O的合成
参考化合物Tap-18H的合成方案,可商购的4-硝基苯基乙醛酸与N-甲基哌嗪缩合,其使用PCl5,或DMF中的EDCI和NiPr2Et,或CH2Cl2中的2-氯-4,6-二甲氧基-1,3,5-三嗪,且N-甲基吗啉作为偶联剂,以生成所需的酮酰胺。在典型的步骤中,在2-氯-4,6-二甲氧基-1,3,5-三嗪(5mmol)在CH2Cl2(20ml)中的溶液中,在0–5℃连续搅拌下添加N-甲基吗啉(15mmol)。30–40分钟后形成白色悬浮液,且向该混合物添加CH2Cl2(10ml)中的4-硝基苯基乙醛酸,导致形成澄清溶液。搅拌该混合物1小时后,在室温添加N-甲基哌嗪(5mmol)。反应完成后(TLC,10分钟),将混合物用10%NaHCO3水溶液(2×10ml)洗涤,然后用H2O(3×10ml)洗涤。有机层用无水硫酸钠干燥且在减压下去除溶剂得到粗产物,其进一步通过重结晶或柱色谱法(石油醚:乙酸乙酯=8:2)纯化。
酮酰胺化合物进一步在THF或DIBAL-H或硼氢化钠的存在下通过0.5当量的LiAlH4还原以产生硝基化合物C。[B.P.Bandgar和S.S.Pandit,Tetrahedron Letters 44(2003)3855–3858]
硝基化合物C通过用SnCl2处理或用Pd/C(10%w/w)作为催化剂在甲醇中在室温下催化氢化约6-11小时而还原为苯胺化合物I,产率为65-81%。
它可以通过使用具有RB04-50反应器B的MultiMaxIR系统通过以下程序获得。反应器最初用35ml甲醇、0.03mg 10%Pd/C和0.0252mol硝基化合物C填充,且在反应器中添加氢气达到6.3巴(H2,恒定)的压力。
参考合成化合物M的方案,将Boc-保护的L-缬氨酸用在DCM中的N-羟基琥珀酰亚胺和EDAC-HCl或在DCM中的N-羟基琥珀酰亚胺和EDC处理,得到琥珀酰亚胺酯。使该活化的酯与L-瓜氨酸和CH3CN、H2O、NaHCO3反应,得到Boc-保护的化合物M。
参考化合物Tap-18H的合成方案,通过DCC/HOBt在DMF中在室温下将苯胺化合物I与Boc-保护的化合物M偶联32小时,得到化合物N(产率78-82%),或与PS-碳二亚胺反应偶联,在该反应中,在两当量的PS-碳二亚胺和1.7当量的DCM中的HOBt的存在下,从100mg的化合物M与1.5当量的苯胺化合物I开始进行化合物N的合成,持续24小时。通过LC/MS分析显示具有所需质量和约50-60%转化率的峰。
偶联的产物化合物N然后与4-硝基氯甲酸苯基酯在DCM中的2,6-二甲基吡啶的存在下在室温反应8小时,以得到碳酸酯化合物P,LC/MS显示具有所需质量的峰。
在DMF中的HOAt和Et3N的存在下用单甲基多拉司他汀10处理碳酸酯化合物P,导致形成化合物Q。
参考化合物O的合成方案,β-丙氨酸用马来酸酐在DMF中处理,且如此得到的酸与N-羟基琥珀酰亚胺(NHS)在DCC偶联下反应,以得到NHS-酯。可商购的t-boc-N-酰胺基-dPEG4-酸中的BOC保护基通过用TFA处理而去除以得到所述胺的TFA盐,将其与之前合成的NHS酯反应。分离如此得到的羧酸且使用EDCI与N-羟基琥珀酰亚胺偶联以得到NHS酯化合物O。
参考化合物Tap-18H的合成方案,化合物Q中的Boc-基团用TFA去除,且游离胺在无水乙腈和NaHCO3中在室温与NHS酯化合物O偶联12-36小时,以制备最终产物Tap-18H,产率为35-45%。
图1显示Tap-18H的NMR谱。
化合物TAP-18Hr1的合成
Tap-18Hr1通过下式合成。图2显示Tap-18Hr1的NMR谱。
化合物TAP-18Hr2的合成
Tap-18Hr2通过下式合成。图3显示Tap-18Hr2的NMR谱。
细胞系
人卵巢癌细胞SKOV-3(ATCC,目录号HTB-77)在补充有10%FBS(HyClone,目录号SH30071.03)、100U/mL青霉素/100μg/mL链霉素(GIBCO,目录号15140)的McCoy's 5A培养基(修饰的)(GIBCO,目录号16600)中培养。人乳腺癌细胞MDA-MB-453(BCRC,目录号60429)在补充有10%FBS(HyClone,目录号SH30071.03)、100U/mL青霉素/100μg/mL链霉素(GIBCO,目录号15140)的Leibovitz's L-15培养基(GIBCO,目录号11415)中培养。人乳腺癌细胞JIMT-1(DSMZ,目录号ACC 589)在补充有10%FBS(HyClone,目录号SH30071.03)、100U/mL青霉素/100μg/mL链霉素(GIBCO,目录号15140)的Dulbecco's MEM培养基(GIBCO,Cat.No.11965)中培养。人胃癌细胞NCI-N87(CCRC,目录号60217)和人T白血病细胞Jurkat(BCRC,Cat.No60424)在补充有10%FBS(HyClone,目录号SH30071.03)、100U/mL青霉素/100μg/mL链霉素(GIBCO,目录号15140)的RPMI培养基1640(GIBCO,目录号22400)中培养。
试剂
DTT和DTPA获自SIGMA-Aldrich(St.Louis,MO)。TCEP获自Acros(Morris Plains,NJ)。DTNB获自Thermo Scientific(Rockford,IL)。磷酸钠、硼酸钠和氯化钠获自J.T.Baker(Center Valley,PA)。半胱氨酸获自Alfa Aesar(Ward Hill,MA)。
抗HER2-半胱氨酸变体的产生
将半胱氨酸残基引入人源化抗HER2抗体(轻链如SEQ ID NO.9且重链如SEQ ID NO.10(对于IgG1)或SEQ ID NO.11(对于IgG4)),使用定向诱变方法。简言之,通过重叠PCR进行诱变。可通过掺入核苷酸改变的引物来引入所需碱基中的具体替换。当引物延伸时,在所得扩增子中产生突变。突变位置和相应的侧翼序列列于下表3中。
表3:半胱氨酸取代的突变体
表达抗HER2-Cys变体的稳定细胞系的产生
在Flp-In CHO细胞(Invitrogen,目录号R708-07)中稳定表达和产生抗HER2半胱氨酸(抗HER2-Cys)变体(表1)。将半胱氨酸取代的抗体变体的DNA序列插入pcDNA5/FRT载体(Invitrogen,目录号V6010-20)中,并按照供应商提供的标准程序与pOG44(Invitrogen,Cat.No V6005-20)共转染。收集建立的细胞系的培养物上清液,并用蛋白A琼脂糖珠子(GE Healthcare,目录号17-5280-04)纯化。纯化的蛋白质用SDS-PAGE和尺寸排阻色谱法分析以确保抗体的质量。
抗HER2-IgG1抗体的常规缀合
在37℃将抗HER2-IgG1(轻链如SEQ ID NO.9,重链如SEQ ID NO.10)抗体用约0.025M硼酸钠pH 8、0.025M NaCl、1mM DTPA中的1.55当量的TCEP还原2小时。对于1.0mg/mL溶液,使用在280nm处的吸光度值为1.48来定量蛋白质浓度,并且使用分子量145,532g/mol测定摩尔浓度。通过用DTNB滴定来测定产生的mAb-半胱氨酸硫醇的浓度。通常得到3.0硫醇/mAb。将部分还原的抗体用1.2摩尔的马来酰亚胺己酰基药物/mAb半胱氨酸硫醇或马来酰亚胺-药物(Tap18Hr1,Tap-18Hr1)/mAb-半胱氨酸硫醇烷基化。烷基化反应在4℃下进行12~16小时。半胱氨酸(1mM终浓度)用于淬灭任何未反应的过量的马来酰亚胺基己酰基-药物或马来酰亚胺-药物。Tap18Hr1缀合混合物首先用结合缓冲液、10mM磷酸钠、10mM NaCl、5%DMSO pH7.0稀释5倍,并以每20mg缀合的抗体1mL羟基磷灰石(称为抗HER2/Tap18Hr1)的负载能力施加到羟基磷灰石柱(Macroprep陶瓷I型40μm,BioRad,Hercules,CA)。该柱预先用5倍柱体积的结合缓冲液平衡。施加样品后,用3倍柱体积的结合缓冲液洗涤柱,然后用5倍柱体积的10mM磷酸钠、10mM NaCl,pH7.0平衡。然后用200mM磷酸钠、10mM NaCl,pH 7.0洗脱结合ADC。洗脱后,使用HiPrepTM 26/10脱盐柱(任选)将缓冲液更换为Dulbecco's磷酸盐缓冲盐水。
抗HER2-Cys变体的位点特异的缀合
为了在所引入的半胱氨酸上特异性缀合连接基有效负载物(payload)Tap18Hr1(Tap-18Hr1),使用还原/氧化步骤。为了除去在培养条件下可能发生的引入的半胱氨酸位点上的半胱氨酸或谷胱甘肽,首先在37℃下在含有1mM DTPA(Sigma-Aldrich,目录号D6518)的PBS(目录号:21600-069)中用10~15倍摩尔过量的TCEP(Acros Organics,目录号363830100)处理抗HER2-Cys变体2~5小时。在除去过量的TCEP后,然后将抗体用相对抗体20-70倍摩尔过量的脱氢抗坏血酸(DHA)(Sigma-Aldrich,Cat.No:261556)在室温下再次氧化3~5小时或在4℃再次氧化3~16小时,以确保链间二硫键的重新形成。将样品的缓冲液交换成PBS。然后加入马来酰亚胺连接的药物有效负载物(Tap18Hr1)以与加工的抗体上的游离巯基反应。用N-乙酰基-L-半胱氨酸(Sigma-Aldrich,Cat.No:A7250)淬灭过量的有效负载物,并使用CHT陶瓷羟基磷灰石(Bio-Rad,目录号157-0040)纯化缀合的抗体。
通过反相HPLC分析确定药物抗体比例(DAR)
HPLC分析之前,缀合物样品用6M胍盐酸盐和20mM DTT在50℃加热下处理15分钟。将100μg处理的缀合物样品施加至PLRP-S柱(2.1x 150mm,8μm,Agilent)。流速为0.8mL/min且柱温度为80℃。溶剂A为mini Q水中的0.05%三氟乙酸且溶剂B为乙腈中的0.04%三氟乙酸。该方法由以下组成:等度25%B 3ml,25ml线性梯度至50%B,2ml线性梯度至95%B,1ml线性梯度至25%B,和等度25%B 2ml。峰归属使用未缀合的抗体(L0和H0)进行。H1和H2通过其洗脱时间和UV谱分配(A248/280比例随着药物负载增加)。
抗HER2抗体和Tap18Hr1缀合物与癌细胞的结合
将1×105细胞接种在v-底96孔板中的每个孔中,并与100μl所示浓度的未缀合的Abs或ADC孵育。在4℃下孵育60~90分钟后,将细胞用200μl FACS缓冲液(1×含有1%FBS的PBS)洗涤,用在FACS缓冲液中的100μl的1μg/ml山羊F(ab')2-抗人IgG(H+L)-RPE(Southern Biotech,目录号2043-09)染色,然后在4℃孵育30~60分钟。用FACS缓冲液洗涤细胞一次,并通过流式细胞术(BD LSR,BD Life Sciences)进行分析。
体外细胞毒性测试:WST-1测试
在96孔微量滴定板上,将SKOV-3细胞以每孔5x103个细胞接种,将MDA-MB-453和JIMT-1以每孔2x104个细胞接种,将NCI-N87以每孔4x104个细胞接种,并将Jurkat细胞以每孔2.5x104个细胞接种。以最终体积200μL/孔以所示浓度一式六份添加抗HER2/Tap18Hr1或未缀合抗体。然后将MDA-MB-453细胞在37℃和0%CO2下孵育96小时,在48小时更新等量培养基。SKOV-3、JIMT-1、NCI-N87和Jurkat细胞在37℃和5%CO2下孵育68-72小时。孵育后,根据制造商的说明书,通过细胞增殖试剂WST-1(Roche,目录号11644807001)检测细胞活力。简言之,在孵育结束时,取出100μL培养基,并加入10μL/孔的WST-1。在最佳显色后(当未处理对照的OD450≥1时,通过分光光度计(Molecular Devices(Sunnyvale,CA),VERSAmax酶标仪)测量450nm处的吸光度(OD450值)。获得一式两份的平均值,且减去背景(介质对照)。然后根据下式使用所得OD450值计算%抑制:[OD450溶剂-OD450样品]/[OD450溶剂]*100。
癌症异种移植模型中的ADC处理
用抗HER2/Tap18Hr1处理的SKOV-3
为了建立皮下异种移植模型,将含有25%高浓度基质胶(BD Biosciences,目录号354248)的100μL PBS中的1x107SKOV-3细胞植入6周龄雌性C.B-17SCID小鼠的右胁腹(Lasco,Taipei,Taiwan)。在肿瘤细胞接种后大约2小时(标记为第1天)以100μL 3mg/kg静脉内注射抗HER2/Tap18Hr1。每周用测径器在两个垂直维度上测量肿瘤体积一次或两次,并根据公式(0.52*长度*宽度*宽度)计算。
用抗HER2/Tap18Hr1处理的MDA-MB-453
为了建立皮下异种移植模型,将含有50%高浓度基质胶(BD Biosciences,目录号354248)的150μL PBS中的1x107MDA-MB-453细胞植入7周龄雌性C.B-17SCID小鼠的右胁腹(Lasco,Taipei,Taiwan)。当平均肿瘤体积达到150mm3,以100μL 3mg/kg静脉内注射抗HER2/Tap18Hr1一次(标记为第1天)。每周用测径器在两个垂直维度上测量肿瘤体积两次,并根据公式(0.52*长度*宽度*宽度)计算。
用抗HER2/Tap18Hr1和位点特异的Tap18Hr1-缀合的抗HER2半胱氨酸变体处理的NCI-N87
为了建立皮下异种移植模型,将在100μL PBS中的5x106NCI-N87细胞植入7周龄雌性C.B-17SCID小鼠的右胁腹(Lasco,Taipei,Taiwan)。当平均肿瘤体积达到180mm3,将药物缀合的抗体静脉内注射(标记为第1天)。每周用测径器在两个垂直维度上测量肿瘤体积一次或两次,并根据公式(0.52*长度*宽度*宽度)计算。
用抗HER2/Tap18Hr1和位点特异的Tap18Hr1-缀合的抗HER2半胱氨酸变体处理的JIMT-1
为了建立皮下异种移植模型,将100μL PBS中的5x106JIMT-1细胞植入6周龄雌性C.B-17SCID小鼠的右胁腹(Lasco,Taipei,Taiwan)。当平均肿瘤体积达到100mm3,以100μL 3mg/kg静脉内注射药物缀合的抗体一次(标记为第1天)。每周用测径器在两个垂直维度上测量肿瘤体积两次,并根据公式(0.52*长度*宽度*宽度)计算。
实施例2:基于抗HER2抗体的抗体药物缀合物(ADC)对癌细胞的体外细胞结合活性
抗HER2/Tap18Hr1结合能力
在SKOV-3、NCI-N87、MDA-MB-453和Jurkat细胞中评价抗HER2裸抗体和Tap18Hr1-缀合的抗体的结合能力。表4中的数据显示测试样品显著结合人HER2-表达细胞系(NCI-N87、SKOV-3和MDA-MB-453),但不结合不表达人HER2的Jurkat。此外,裸抗体和药物-缀合的抗体都以相当的平均荧光强度(MFI)结合这些细胞。这些结果表明抗HER2/Tap18Hr1保持裸抗体的抗原反应性并有效地结合表达HER2的细胞。
表4:抗HER2-IgG1与癌细胞的结合
在乳腺JIMT-1、胃NCI-N87和卵巢SKOV-3癌细胞中Tap18Hr1-缀合的抗HER2半胱氨酸变体的结合能力
在乳腺JIMT-1(表5-7),胃NCI-N87(表8)和卵巢SKOV-3(表9)癌细胞中评价具有或不具有药物缀合的抗-HER2-IgG1Cys变体的结合能力。表5-9中的数据显示抗HER2-IgG1半胱氨酸变体与抗HER2-IgG1Ab相比同等地结合所有测试的癌细胞。此外,位点特异性缀合的抗HER2-IgG1ADC还保留了抗原反应性,除了显示比其它变体略低的亲和力的抗HER2-S442C-IgG1/Tap18Hr1。
表5.抗HER2-IgG1变体与JIMT-1细胞的结合
*ND:未测定。
表6.抗HER2-IgG1变体与JIMT-1细胞的结合
*ND:未测定。
表7.抗HER2-IgG1变体与乳腺JIMT-1癌细胞的结合
*ND:未测定。
表8.抗HER2-IgG1变体与胃NCI-N87癌细胞的结合
*ND:未测定。
表9.抗HER2-IgG1变体与卵巢SKOV-3癌细胞的结合
*ND:未测定。
在乳腺JIMT-1(表10-11)、胃NCI-N87(表12)和卵巢SKOV-3(表13)癌细胞中评价具有或不具有药物缀合的抗HER2-IgG4p Cys变体的结合能力。表10-13中的数据显示抗HER2-IgG4p半胱氨酸变体的结合与抗HER2-IgG4p抗体的结合相当,但与抗HER2-IgG1抗体的结合相比较低(表5-9),表明降低的结合活性归因于IgG4同种型而不是半胱氨酸突变。总的来说,位点特异性缀合的抗HER2-IgG4p ADC保留了抗原反应性。与IgG1变体相似,抗HER2-S442C-IgG4p也显示出比其它变体稍低的亲和力。
表10.抗HER2-IgG4p半胱氨酸变体与JIMT-1细胞的结合
*ND:未测定。
表11.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸变体与乳腺JIMT-1癌细胞的结合
*ND:未测定。
表12.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸变体与胃NCI-N87癌细胞的结合
*ND:未测定。
表13.抗HER2-IgG1、抗HER2-IgG4p和抗HER2-IgG4p半胱氨酸变体与卵巢SKOV-3癌细胞的结合
*ND:未测定。
实施例3:基于抗HER2抗体的抗体药物缀合物(ADC)对癌细胞的体外细胞毒性作用
在HER2阳性癌细胞系(NCI-N87,SKOV-3,MDA-MB-453和JIMT-1)和HER2阴性细胞系(Jurkat)中评价常规缀合的抗HER2抗体(抗HER2/Tap18Hr1)的体外细胞毒活性。也平行测试了裸抗体的细胞毒性。在5和1.25μg/mL,尽管抗HER2裸抗体可以诱导NCI-N87(胃癌细胞)和MBA-MD-453(乳腺癌细胞)中的细胞毒性,但常规缀合的抗HER2甚至更有效(表14)。对于JIMT-1(乳腺癌细胞)和SKOV-3(卵巢癌细胞),只有药物缀合的抗HER2可以在5和1.25μg/mL引起超过50%的生长抑制(表14和15)。在HER2阴性细胞系Jurkat中没有观察到毒性。这些结果表明Tap18Hr1缀合的抗HER2递送细胞毒性药物到具有抗原特异性的靶癌细胞。
表14.体外细胞毒性活性
阴性值表明没有检测到细胞毒性。
表15.体外细胞毒性活性
还在NCI-N87、JIMT-1和SKOV-3细胞中评价了位点特异性缀合的抗HER2-Cys变体的体外细胞毒性活性。表16和17显示了抗-HER2半胱氨酸变体的测试ADC的细胞毒性测定结果。在5和1.25μg/mL,位点特异性ADC有效杀死HER2阳性癌细胞(NCI-N87,JIMT-1和SKOV-3),但不在HER2阴性细胞系(Jurkat)中杀死细胞。尽管是稍低的结合,但抗HER2-S442C/Tap18Hr1在抗原表达细胞中诱导与其他半胱氨酸变体相似的细胞毒性程度。这些结果表明,位点特异性抗HER2ADC可以递送细胞毒性药物至具有抗原特异性的靶癌细胞。
表16.Tap18Hr1缀合的抗HER2-Cys变体的体外细胞毒性活性
表17.Tap18Hr1缀合的抗HER2-Cys变体的体外细胞毒性活性
实施例4:用常规缀合的抗HER2处理的SKOV-3异种移植物
在体内评价抗HER2/Tap18Hr1对卵巢癌细胞SKOV-3的功效。在肿瘤细胞接种后大约两小时(标记为第1天),用媒介物(PBS,100μL)或100μL的300mg/kg单剂量ADC以静脉内处理小鼠。由于包括基质胶(Matrigel)的接种体积,第1天的肿瘤大小记录为100mm3。注射的基质胶到第15天被吸收,同时肿瘤建立并在媒介物组中稳定生长(图4)。在第17天用抗HER2/Tap18Hr1处理抑制肿瘤生长,并且该组中的所有小鼠(5/5)显示自第27天以来不可检测的肿瘤。由于两组的体重稳定地增加,观察不到毒性。实验表明,使用单一注射,抗HER2/Tap18Hr1可有效抑制SCID小鼠中移植的抗原阳性肿瘤的生长。
实施例5:用常规缀合的抗HER2处理的MDA-MB-453异种移植物
在体内评价抗HER2/Tap18Hr1对乳腺癌细胞MDA-MB-453的功效。当平均接种的肿瘤大小达到~150mm3时,用PBS(媒介物,100μL)或100μL的3mg/kg单剂量ADC以静脉内处理小鼠。抗HER2/Tap18Hr1组在第8天显示肿瘤消退,从第11天起,平均肿瘤大小进一步降低至<50mm3(图5)。在研究结束时,6只小鼠中的3只显示完全肿瘤消退。两组小鼠体重均稳定增加。数据表明,抗HER2/Tap18Hr1可以有效抑制SCID小鼠中移植的抗原阳性肿瘤的生长。
实施例6:用Tap18Hr1常规缀合的抗HER2和位点特异的缀合的抗HER2-Cys变体处理的NCI-N87异种移植物
在体内对胃癌细胞NCI-N87评价常规缀合的抗HER2/Tap18Hr1的功效。当平均肿瘤大小达到~180mm3时,用PBS(媒介物,100μL)或ADC(3或5mg/kg,100μL)静脉内处理小鼠两次(标记为第1天和第22天)。尽管媒介物组的肿瘤生长并在第15天接近500mm3(图6),但抗HER2/Tap18Hr1组在第5天显示延迟的肿瘤生长,从第19天开始,平均肿瘤大小进一步降低至<30mm3。在研究结束时,大多数肿瘤在两个ADC治疗组中低于10mm3。小鼠体重在三组中稳定增加。数据表明,抗HER2/Tap18Hr1可以有效抑制SCID小鼠中移植的抗原阳性肿瘤的生长。
在体内对胃癌细胞NCI-N87评价了位点特异性缀合的抗HER2-Cys变体的功效。当肿瘤大小的平均值达到~180mm3时,用PBS(媒介物,100μL)或具有相等药物剂量(9.7μg/kg Tap18Hr1在100μL中)的ADC静脉内处理小鼠(标记为第1天)。如图7所示,与媒介物组相比,所有位点特异性缀合的变体处理的小鼠显示显着延迟的肿瘤生长。体重在ADC治疗组中保持不变,并且由于肿瘤的重量,在媒介物组中轻微增加。数据证明,通过单次注射,位点特异性缀合的抗HER2-Cys变体可有效抑制在SCID小鼠中移植的抗原阳性肿瘤的生长。
实施例7:用Tap18Hr1常规缀合的抗HER2和位点特异的缀合的抗HER2-Cys变体处理的JIMT-1异种移植物
在体内针对已知的赫赛汀抗性乳腺癌细胞JIMT-1评价常规缀合的抗HER2/Tap18Hr1和位点特异性缀合的抗HER2-Cys变体的功效。当平均肿瘤大小达到~100mm3时,用PBS(媒介物对照,100μL)或ADC(3mg/kg,在100μL中)静脉内处理小鼠一次(标记为第1天)。如图8所示,与媒介物组相比,所有ADC治疗组显示自第7天以来显着延迟的肿瘤生长。小鼠的体重在所有组中稳定增加。数据表明,单次给药,不仅常规抗HER2/Tap18Hr1而且位点特异性缀合的抗HER2-Cys变体都可以有效抑制在SCID小鼠中移植的HER2阳性肿瘤的生长。
- 还没有人留言评论。精彩留言会获得点赞!