光活化电子供体-受体复合物的烷基羧化物的脱羧及脱羧Giese自由基加成反应的制作方法
本发明属于有机化学领域,具体地,涉及一种光活化电子供体-受体复合物的烷基羧化物的脱羧及脱羧giese自由基加成反应。
背景技术:
脂肪族羧酸类物质具有易于操作、产物丰富、环境友好等优良特性,能够在绿色化学合成中被广泛应用。基于此,以脂肪族羧酸类物质作为反应物的烷基羧化物脱羧及脱羧giese自由基加成反应得到集中的发展。目前,主要用于促进羧化物脱羧进而形成碳-碳键结构的方法主要是:在高温的反应条件下,利用过渡金属复合物作为催化剂,催化反应物发生反应。然而,用于以上用途的催化剂往往价格昂贵,大大地提高了生产中的原料成本,降低生产的经济效益。另外,混合在目标产物中的催化剂往往具有较高的毒性且难以与目标产物分离,厂家必须为了分离目标产物中的催化剂而投入大量的技术成本,同时,残存在目标产物中无法被分离的催化剂也会降低目标产物的纯度,影响产品的品质。例如,通过barton脱羧反应得到的脱羧产物中不可避免地混有锡化物和硫醇,这些物质具有较高毒性和刺激性气味,为产物引入安全隐患,为了从产物中分离这些物质所投入的费用占据着产品产出成本中不少的比例。因此,为了响应绿色环保可持续发展的理念以及提高产品的经济效益和安全性,迫切地需要开发一种无需催化剂的羧化物脱羧反应方法。
技术实现要素:
本发明的目的在于提供一种光活化电子供体-受体复合物的烷基羧化物的脱羧及脱羧giese自由基加成反应,以在无需催化剂的条件下实现羧化物的脱羧及脱羧后重新形成碳-碳键的方法。
根据本发明的一个方面,提供一种光活化电子供体-受体复合物的烷基羧化物的脱羧及脱羧giese自由基加成反应:在溶液环境中,使
优选地,ra选自:
优选地,ra选自:
优选地,蓝紫光的波长范围为427–467nm。
优选地,蓝紫光的波长为456nm。
优选地,溶液环境的溶剂选自n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
优选地,溶剂为n,n-二甲基乙酰胺。
优选地,惰性保护气氛为氩气气氛。
优选地,烷基n-(酰氧基)苯邻二甲酰亚胺类化合物和汉斯酯的当量比为1:1.5。
优选地,烷基n-(酰氧基)苯邻二甲酰亚胺类化合物和烯基类化合物的当量比为1:1.5。
上述反应过程不需要使用催化剂,只需要在蓝紫光的活化下即可启动反应,并且使反应物在反应的过程中保持良好的反应活性。
然而,电子供体-受体复合物rae…he并不是一种稳定的组合物,本发明利用n,n-二甲基甲酰胺(dmf)或n,n-二甲基乙酰胺(dma)作为溶剂,为反应物提供溶液反应环境,能够提高rae…he在溶液中的稳定性,避免其在被光活化前发生解离,显著地提高了目标产物的得率。然而,物质溶解在不同溶剂中往往对应不同的光吸收峰,而本发明涉及的反应物的蓝紫光吸收率随着波长的变化存在极大的差异,因此,利用不同溶剂构建的反应体系对激发光的敏感度存在显著的区别。本发明根据反应物本身以及其在不同溶剂中的光吸收特性,对激发光的波长限定在一定范围内,保证反应物能够被有效地活化,使脱羧反应能够顺利进行,并且获得较高的产物得率。
另外,本发明还通过为反应提供合适的保护气氛、限定适当的反应物当量比等反应条件,更好地促进反应物向目标产物转化,提高目标产物得率,降低副产品的产出。
由于上述过程不需要催化剂的参与,从而避免了催化剂的使用为产物引入有毒的、难以分离的杂质,有效地提高了产品的纯度和安全性,也削减了为了购买催化剂而产生的昂贵原料投入以及为了将产物与催化剂分离而产生的设备投入和时间投入,极大地提高了产品的生产效益。
附图说明
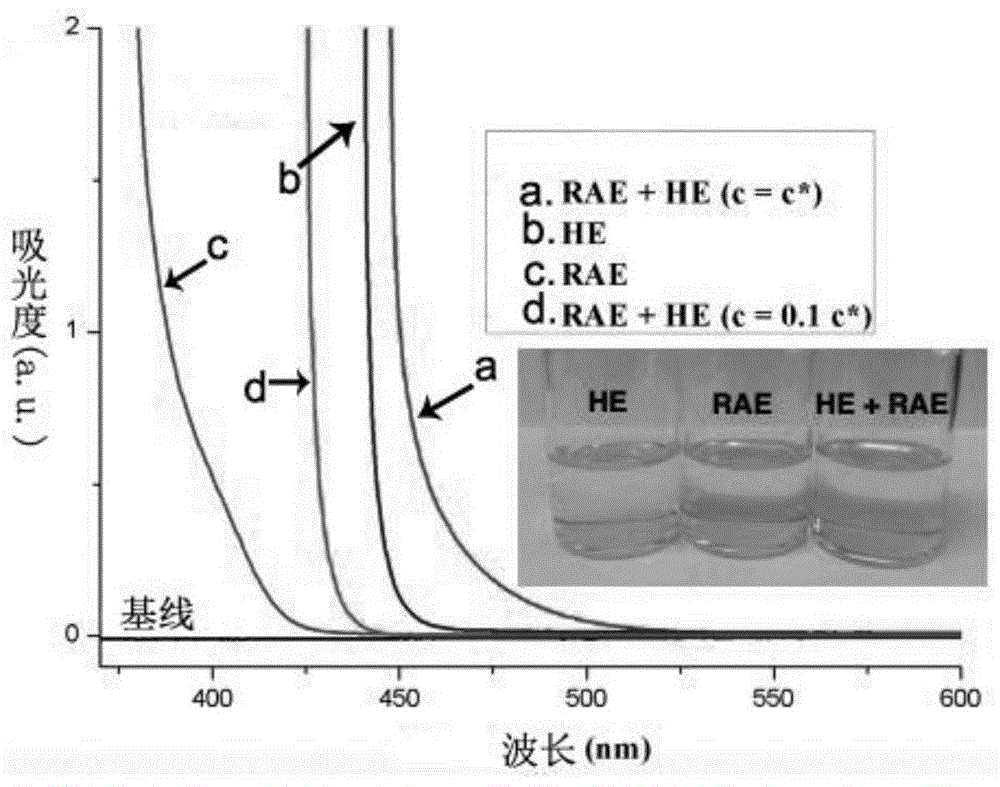
图1为实施例2中设置的溶剂为dma形成的溶液的紫外-可见吸收光谱图,其中:(a)rae的dma溶液(c=c*),(b)he的dma溶液(c=c*),(c)he+rae的dma溶液(c=c*),(d)he+rae的dma溶液(c=0.1c*);
图2为实施例2中设置的溶剂为dcm形成的溶液的紫外-可见吸收光谱图,其中:(a)rae的dcm溶液(c=c*),(b)he的dcm溶液(c=c*),(c)he+rae的dcm溶液(c=c*),(d)he+rae的dcm溶液(c=0.1c*);
图3为实施例1中产物1的1hnmr谱图;
图4为实施例1中产物1的13cnmr谱图;
图5为实施例5中产物2的1hnmr谱图;
图6为实施例5中产物2的13cnmr谱图;
图7为实施例5中产物3的1hnmr谱图;
图8为实施例5中产物3的13cnmr谱图;
图9为实施例5中产物4的1hnmr谱图;
图10为实施例5中产物4的13cnmr谱图;
图11为实施例5中产物5的1hnmr谱图;
图12为实施例5中产物5的13cnmr谱图;
图13为实施例5中产物6的1hnmr谱图;
图14为实施例5中产物6的13cnmr谱图;
图15为实施例5中产物7的1hnmr谱图;
图16为实施例5中产物7的13cnmr谱图;
图17为实施例5中产物8的1hnmr谱图;
图18为实施例5中产物8的13cnmr谱图;
图19为实施例5中产物9的1hnmr谱图;
图20为实施例5中产物9的13cnmr谱图;
图21为实施例5中产物10的1hnmr谱图;
图22为实施例5中产物10的13cnmr谱图;
图23为实施例5中产物11的1hnmr谱图;
图24为实施例5中产物11的13cnmr谱图;
图25为实施例5中产物12的1hnmr谱图;
图26为实施例5中产物12的13cnmr谱图;
图27为实施例5中产物13的1hnmr谱图;
图28为实施例5中产物13的13cnmr谱图。
具体实施方式
为了使本技术领域的人员更好地理解本发明方案,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。
下列实施例中需要用到的主要仪器如下:schlenk管、buchi旋转蒸发器、shimadzugc-2014气相色谱仪、brukeravance400核磁共振波谱仪、brukeravance400核磁共振波谱仪、装配有apci离子源的thermoltqorbitrapxl高分辨质谱仪、uv-3600紫外-可见吸收光谱仪。
除了特别说明外,以下实施例中涉及的脱羧以及giese自由基加成反应均在充满氩气气氛的干燥schlenk管内进行。所使用的试剂均购自阿达玛斯试剂有限公司(adamas)、梯希爱(上海)化成工业发展有限公司(tci)或西格玛奥德里奇(上海)贸易有限公司(aldrich)。下列实施例中采用的光反应实验器材均由anhuikemimachinerytechnologyco.,ltd提供。
在常温下,利用brukeravance400核磁共振波谱仪获取1h-nmr和13c-nmr的谱图数据。1h-nmr中的数据含义如下:化学位移(ppm,scale),谱峰多重性(s=singlet,d=doublet,t=triplet,q=quartet,m=multipletand/ormultipletresonances,br=broad),耦合常数(hz),integration。13c-nmr中的数据含义如下:化学位移(ppm,scale),谱峰多重性,耦合常数(hz)。
以下实施例中涉及的rae的合成方法如下:
实施例1自由基时钟实验
1.主要所需药品
二氯甲烷(dcm)、n,n-二甲基乙酰胺(dma)。
2.数据采集
薄层色谱法(tlc)、气相色谱-质谱联用仪(gc-ms)、核磁共振波谱仪(nmr)
3.自由基时钟实验反应步骤
3.1向配有搅拌器的schlenk管中投入
3.2向配有搅拌器的schlenk管中投入
本实施例设置的自由基时钟实验的反应物反应条件及目标生产产物如下:
4.实验结果:在存在作为自由基清除剂的tempo的反应体系中得到的混合物中,未发现具有上述目标产物(标记为产物1)结构的产品。而在不存在tempo的反应体系中得到的产物的的结构符合上述目标产物结构,得到了40.8mg的目标产物,产率高达88%;产物1的nmr谱图如图3和图4所示,其1hnmr和13cnmr数据如下:
1hnmr(400mhz,cdcl3)δ7.38(t,j=7.9hz,2h),7.23(t,j=7.4hz,1h),7.09(d,j=7.6hz,2h),2.63-2.51(m,2h),1.80-1.70(m,7h),1.38-1.15(m,4h),1.04-0.89(m,2h).
13cnmr(101mhz,cdcl3)δ172.6,150.7,129.3,125.6,121.5,37.2,32.9,32.3,32.0,26.5,26.2.。
由此,给出了上述反应历程为自由基历程的有力证据。
实施例2克级规模实验
本实施例将实施例1中3.2部分所开展的实验扩大规模至克级规模实验。向配有搅拌器的schlenk管中投入
反应物反应条件及目标生产产物如下:
在本实施例开展的克级规模实验中,得到1.13g目标产物,得率高达81%。
实施例3
1.实验设置方式
1.1分别将实施例1采用的he和rae(即,
1.2分别将实施例1采用的he和rae(即,
2.数据获取方式
利用紫外-可见吸收光谱仪测试上述溶液的吸收光谱。
3.测试结果
测试结果如图1和图2所示。如图1所示,与分别将he和rae溶于dma溶液相比,将he和rae同时溶于dma以模拟实施例1中的反应条件的he+rae的dma溶液(c=c*)的吸收光谱峰产生较大距离的红移,he+rae的dma溶液(c=c*)的吸收峰对应溶液中形成的eda复合物,其吸收峰延展至大于500nm的可见光区域。同时,反应物浓度也是影响吸收峰的重要因素。从图1可知,与he+rae的dma溶液(c=c*)的吸收峰相比,he+rae的dma溶液(c=0.1c*)的吸收峰发生显著的蓝移,产生这种现象的原因是,稀释的过程抑制了eda复合物的形成。此外,基于为反应物提供溶液反应环境的溶剂能够对形成的eda复合物的稳定性造成重要的影响,因此,溶剂是影响反应进行效果的关键因素。对比图1和图2展示的吸收光谱图,与he+rae的dma溶液(c=c*)的吸收峰相比,he+rae的dcm溶液(c=c*)的吸收峰发生明显的蓝移,其吸收峰位于波长小于500nm的光谱范围。
实施例4
本实施例以以下对照处理方式为依据设置若干处理组,除特别的变量说明外,每个处理组的处理方式均与对照处理方式严格保持一致。反应式如下:
在本实施例中,上述反应式中的rae指
对照处理方式:
向配有搅拌器的schlenk管中投入
各处理组的变量设置及目标产物的产量如表1所示。
表1本实施例各处理组设置方式对目标产物得率的影响
根据处理2对应的产物得率,对照处理方式中建立的反应条件同样适用于
通过对比处理1、处理3–6各自对应的产品得率可知,溶剂是反应成功的关键因素。因为溶剂与在其中形成的、具有光活性的组合物产生弱非共价键相互作用,从而影响该组合物的稳定性。从表1中展示的数据可知,dma为本反应最合适的溶剂。
通过对比处理1、处理7–9各自对应的产品得率,利用其它的电子供体替代he与其它反应物进行反应,都会大大地降低目标产物的得率。而根据处理14对应的数据可知,he的参与是反应能够顺利进行的必要条件。而根据处理15和16,he与其它反应物的投料当量比也能够在一定程度上对目标产品的得率构成影响。
根据处理19对应的产品得率,光辐照是反应能够顺利进行的必要条件。通过对比处理1、处理10–13各自对应的产品得率,采用390nm的紫光为反应物提供辐照,将显著地降低目标产物得率。而将辐照光的波长位于427–467nm的光谱范围内,在该辐照条件下,能够使目标产物的得率保持在80%以上。
根据处理17,可以证明该反应体系对水的敏感度较低,水的加入不会对产品得率带来显著的影响。然而,根据处理18,氩气保护气氛对反应的顺利进行也十分重要,当将反应物暴露于没有保护气氛的空气下反应,几乎无法检测到目标产物。
实施例5
选择
向配有搅拌器的schlenk管中投入
表2为本实施例采用的不同种类的rae作为反应物参与反应对应得到的目标产物及其得率。
表2rae种类对目标产物类型及其产率的影响
表2中列举的产物的1hnmr和13cnmr数据如下:
产物2:
1hnmr(400mhz,cdcl3)δ7.39-7.21(m,5h),5.32-5.22(m,2h),5.04(s,2h),2.28(t,j=7.6hz,2h),2.02-1.86(m,4h),1.56(dd,j=14.4,7.2hz,2h),1.24-1.19(m,24h),0.81(t,j=6.8hz,3h).
13cnmr(101mhz,cdcl3)δ173.6,136.1,134.2,128.4,128.1,66.0,34.2,33.9,32.7,29.8,29.7,29.6,29.5,29.4,29.3,29.2,29.1,29.0,28.7,28.1,24.9,22.6,14.1.。
产物3:
1hnmr(400mhz,cdcl3)δ7.32-7.20(m,5h),5.04(s,2h),3.33(t,j=6.9hz,2h),2.28(t,j=7.5hz,2h),1.85-1.73(m,2h),1.56(dd,j=14.3,7.2hz,2h),1.33(dd,j=14.1,6.8hz,2h),1.25-1.14(m,j=6.0hz,14h).
13cnmr(101mhz,cdcl3)δ173.6,136.1,134.2,128.4,128.1,66.0,34.2,33.9,32.7,29.5,29.4,29.3,29.2,29.1,28.7,28.1,24.9.。
产物4:
1hnmr(400mhz,cdcl3)δ7.36-7.20(m,5h),5.04(s,2h),3.44(t,j=6.7hz,2h),2.29(t,j=7.5hz,2h),1.75-1.53(m,4h),1.46-1.20(m,4h).
13cnmr(101mhz,cdcl3)δ173.4,136.0,134.2,128.5,128.1,66.0,44.8,34.0,32.3,28.2,26.4,24.6.。
产物5:
1hnmr(400mhz,cdcl3)δ7.42-7.29(m,5h),5.12(s,2h),2.36(t,j=7.5hz,2h),2.17(td,j=7.0,2.6hz,2h),1.93(t,j=2.7hz,1h),1.67(dd,j=15.0,7.5hz,2h),1.58-1.42(m,2h),1.46-1.28(m,4h).
13cnmr(101mhz,cdcl3)δ173.5,136.0,128.5,128.1,117.7,84.5,68.2,66.0,34.2,28.5,28.3,28.2,24.7,18.2.。
产物6:
1hnmr(400mhz,cdcl3)δ7.40-7.28(m,5h),7.26-7.21(m,2h),7.12(dd,j=11.6,8.5hz,2h),5.12(s,2h),2.55-2.36(m,3h),1.92-1.72(m,3h),1.67-1.57(m,6h),1.45-0.97(m,2h).。
产物7:
1hnmr(400mhz,cdcl3)δ7.49-7.28(m,10h),5.12(s,2h),4.84-4.55(m,1h),3.86-3.55(m,1h),3.09-2.56(m,2h),2.40(t,j=7.6hz,2h),1.79-1.38(m,5h),1.28-1.03(m,2h).
13cnmr(101mhz,cdcl3)δ173.3,170.3,136.3,135.9,129.4,128.4,128.3,128.2,126.8,123.5,66.3,47.8,42.3,35.5,31.5,31.2.。
产物8:
1hnmr(400mhz,cdcl3)δ7.50-7.18(m,5h),5.04(s,2h),2.40-2.10(m,2h),1.56-1.51(m,2h),1.22-1.15(m,9h),0.82-0.74(m,6h).
13cnmr(101mhz,cdcl3)δ173.9,136.1,128.4,128.2,128.1,66.0,38.3,32.4,31.7,28.7,28.1,25.5,23.0,14.0,10.6.。
产物9:
1hnmr(400mhz,cdcl3)δ7.35-7.21(m,5h),5.04(s,2h),2.37-2.23(m,2h),1.77-1.34(m,9h),1.08-0.90(m,2h).
13cnmr(101mhz,cdcl3)δ173.7,136.1,128.5,128.2,128.1,66.0,39.6,33.6,32.3,31.0,25.0.。
产物10:
1hnmr(400mhz,cdcl3)δ7.40-7.19(m,5h),5.03(s,2h),2.34-2.13(m,2h),1.53(dd,j=10.6,6.3hz,2h),1.41-1.28(m,5h),1.32-1.14(m,5h),0.77(s,3h).
13cnmr(101mhz,cdcl3)δ174.4,136.0128.4,128.2,128.1,66.1,37.4,36.6,32.2,28.9,26.3,24.3,21.9.。
产物11:
1hnmr(400mhz,cdcl3)δ7.34-7.22(m,5h),5.04(s,2h),2.36-2.19(m,2h),1.52-1.48(m,2h),0.82(s,9h).
13cnmr(101mhz,cdcl3)δ174.2,136.0,128.5,128.2,128.1,66.138.5,30.1,30.0,28.9.。
产物12:
1hnmr(400mhz,cdcl3)δ7.35-7.19(m,5h),6.91(d,j=7.4hz,1h),6.63-6.49(m,2h),5.03(s,2h),3.81(t,j=6.3hz,2h),2.33-2.16(m,5h),2.09(s,3h),1.73-1.50(m,4h),1.33-1.22(m,2h),0.82(s,6h).
13cnmr(101mhz,cdcl3)δ174.0,156.9,136.3,136.0,130.2,128.4,128.2,128.1,123.4,120.5,111.8,68.3,66.1,37.7,36.2,32.2,29.5,26.7,24.1,21.3,15.7.。
上述产物2-12的nmr谱图如图5-26所示。
选择
向配有搅拌器的schlenk管中投入
采用上述原料及操作步骤得到的目标产物结构为:
1hnmr(400mhz,cdcl3)δ7.41-7.32(m,2h),7.23-7.18(m,1h),7.13-7.01(m,4h),6.85-6.80(m,2h),3.78(s,3h),2.60-2.52(m,4h),1.81-1.61(m,4h),1.50-1.37(m,2h).
13cnmr(101mhz,cdcl3)δ172.2,157.7,150.7,134.5,129.4,129.3,125.7,121.6,113.7,55.2,34.7,34.3,31.3,28.6,24.8.。
根据本实施例的实验结果,当
以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!