一种手性2-芳基丙酸酯的合成方法与流程
本发明属于化学合成的技术领域,涉及到对映体选择性合成2-芳基丙酸酯。
背景技术:
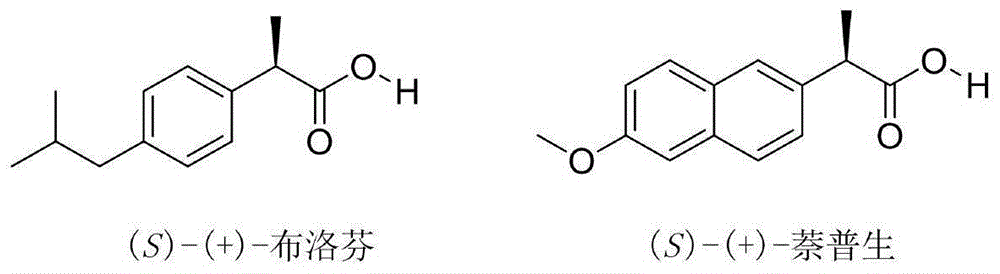
2-芳基丙酸是常见的止痛消炎类药物。其中,布洛芬(ibuprofen)为otc类非甾体抗炎药[1],广泛用于治疗头痛、神经痛、风湿性关节炎和类风湿性关节炎等疾病。其有效成分为(s)-(+)-布洛芬,而(r)-(-)-布洛芬异构体无效。萘普生(naproxen)是pg合成酶抑制剂,多种急性疼痛的镇痛药,有抗炎、解热、镇痛作用,对类风湿性关节炎、骨关节炎、强直性脊椎炎、痛风、运动系统的慢性变性疾病及轻、中度疼痛等,均有肯定疗效,用于偏头痛,紧张性头痛,术后疼痛和与各种妇科手术相关的疼痛的缓解[2]。临床结果表明,(s)-(+)-萘普生的生物活性比(r)-(-)-萘普生的活性高28倍。(s)-(+)-布洛芬和(s)-(+)-萘普生的结构如图1。
外消旋布洛芬的合成包括boots法和bhc法。boots法是以异丁基苯为原料,通过傅克反应生成异丁基苯乙酮,再经darzens反应、氧化反应生成布洛芬[3]。该法存在着路线长、原子经济性差、原料成本高的问题,而且对设备耐腐性能要求高;bhc法是通过1-(4-异丁基苯基)乙酮经还原、醇羰化反应合成布洛芬[4],其中羰化反应采用pd(pph3)2cl2作催化剂。该法原子经济性好,但存在贵金属pd的成本高及毒性、反应需高压才能进行等问题。萘普生的合成方法及存在的问题与布洛芬的相似。
手性(s)-(+)-布洛芬和(s)-(+)-萘普生主要分别通过外消旋布洛芬和萘普生的拆分得到。采用不对称催化法制备这些化合物,可实现高原子经济性反应,减少无效异构体的生成及对人体或环境的影响,具有良好的工业化应用前景。目前的主要方法为过渡金属rh、ru、pd催化芳基乙烯的不对称氢甲酰化[5]或氢酯化反应[6],或1-芳基乙醇的不对称羰基化反应[7],或2-芳基丙烯酸的不对称氢化反应[8,9]。然而,采用的催化剂中过渡金属价格昂贵、需高压,这些因素限制了其工业应用。
参考文献
1.kantor,t.g.,ibuprofen.annalsofinternalmedicine1979,91(6),877-82.
2.todd,p.a.;clissold,s.p.,naproxen.areappraisalofitspharmacology,andtherapeuticuseinrheumaticdiseasesandpainstates.drugs1990,40(1),91-137.
3.于凤丽,赵玉亮,金子林.布洛芬合成绿色化进展.有机化学,2003,11,1198-1204
4.sheldon,r.a.chemtech1994,24,38.
5.francio,g.;leitner,w.,highlyregio-andenantio-selectiverhodium-catalysedasymmetrichydroformylationwithoutorganicsolvents.chem.commun.1999,(17),
1663-1664.
6.cometti,g.;chiusoli,g.p.j.organomet.chem.1982,236,c31.
7.xie,b.h.;xia,c.g.;lu,s.j.;chen,k.j.;kou,y.;yin,y.q.,thefirstasymmetriccarbonylationof1-(6'-methoxy-2'-naphthyl)ethanoltothemethylesterof(s)-naproxen.tetrahedronlett.1998,39(40),7365-7368.
8.monteiro,a.l.;zinn,f.k.;desouza,r.f.;dupont,j.,asymmetrichydrogenationof2-arylacrylicacidscatalyzedbyimmobilizedru-binapcomplexin1-n-butyl-3-methylimidazoliumtetrafluoroboratemoltensalt.tetrahedron-asym.1997,8(2),177-179.
9.张绪穆,陈才友,手性双膦配体及其在不对称氢化及相关反应中的应用,中国专利申请号201510765575.7,2015.11.11.
技术实现要素:
针对以上问题,本发明利用廉价铜为催化剂、硅烷为还原剂,在手性膦配体存在下,还原2-芳基丙烯酸酯,生成手性2-芳基丙酸酯。目标化合物经简单水解,就能得到手性2-芳基丙酸。本发明方法条件温和、操作简单、对环境友好,具有一定的手性选择性。本发明方法得到的2-(4-甲氧基苯基)丙酸酯等可以扩展应用于这两种药物的合成。
本发明的合成方法为:将铜盐、手性膦配体、硅氢化合物(以sih计)、r1oh与2-芳基丙烯酸酯按照一定比例加入到反应瓶中,在反应溶剂中于-50-40℃反应0.25-6h,反应结束后经水解、分液、萃取、洗涤、干燥、柱层析得到目标化合物。
利用不对称催化还原2-芳基丙烯酸酯合成2-芳基取代丙酸酯反应式如下:
其中,ar=ph,4-f-ph,4-cl-ph,4-br-ph,4-i-ph,4-meo-ph,4-me-ph,4-me2chch2-ph,1-nap,2-nap,2-(6-meo-2-nap),2-(6-me-2-nap),2-(6-cl-2-nap)或2-(6-br-2-nap)中的一种;优选4-me2chch2-ph和2-(6-meo-2-nap)。r=me,et,cf3cf2,n-pr,i-pr,n-bu,i-bu,t-bu等c1-c5的烷基,或苯基、苄基、带甲基或乙基的取代苯基、取代苄基。
所述的铜盐、手性膦配体、硅氢化合物(以sih计)、r1oh与2-芳基丙烯酸酯的摩尔比为(0.0005~0.03):(0.0005~0.03):(1~5):(0~4):1。
所述的铜盐为:cuf(pph3)3·2meoh、cu(oac)2·h2o、cux与叔丁醇钠或叔丁醇钾的混合物,其中x为f、cl、i中的一种。为避免叔丁醇钾或钠过量导致消旋化,cux与叔丁醇钾或钠的比例优选在2:1-1:1。理论上,二者比例1:1最好,其中cu(ii)被还原为cu(i)。
所述的硅氢化合物为sih、phsih3、ph2sih2、ph3sih、ph2mesih、(sihme2)2o、et3sih、(meo)3sih、(sihme2)2nh。
所述的r1oh为1-6个碳原子的一级、二级或三级醇。
所述的手性膦配体为有如下结构的膦化合物l1-l6或它们的对映体。
其中,ar'=ph,3,5-me2-ph,3,4,5-(meo)2-ph,4-meo-3,5-(t-bu)2-ph。
所述的反应溶剂为醚类或芳香烃类或卤代烷,包括乙醚、丁醚、四氢呋喃、苯、甲苯、二甲苯、二氯甲烷、1,2-二氯乙烷等。
与现有技术相比较,本发明采用cu催化体系还原2-芳基丙烯酸酯,催化剂cu化合物价格便宜,突破了氢气高压还原和贵金属催化剂的限制。产物经简单水解反应就可得到手性2-芳基丙酸,可作为有效成分生产布洛芬、萘普生等药物。
附图说明
图1(s)-布洛芬与(s)-萘普生的结构;
图2配体的结构;
图3外消旋2-苯基丙酸甲酯的手性gc谱图;
图4不对称催化反应生成的2-苯基丙酸甲酯的手性gc谱图;
图5外消旋2-(4-异丁基苯基)丙酸甲酯的手性hplc谱图;
图6不对称催化反应生成的2-(4-异丁基苯基)丙酸甲酯的手性hplc谱图。
图7不对称催化反应生成的2-(2-氯苯基)丙酸甲酯的手性hplc谱图;
图8不对称催化反应生成的2-(4-甲氧基苯基)丙酸甲酯的手性hplc谱图。
具体实施方式
下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
实施例1
给氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(r,r)-l1(ar'=ph)(2.0mg)和甲苯(2.0ml),室温下搅拌至蓝色溶液。给上述混合物中加入聚(甲基氢硅氧烷)(120μl)、正丁醇(118μl),搅拌5min后加入2-苯基丙烯酸甲酯(0.065g),再搅拌2h。之后,给反应混合物中加入饱和nh4f溶液(4.0ml),继续搅拌60min,分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得到2-苯基丙酸甲酯0.046g,产率70%;经手性gc手性分析对映体组成,ee8%,优势构型为(s)-构型。1hnmr(500mhz,cdcl3)δ7.32–7.20(m,5h),3.66(q,j=7.1hz,1h),3.59(s,3h),1.43(d,j=7.2hz,3h).13cnmr(126mhz,cdcl3)δ175.13,140.67,128.77,127.59,127.26,52.17,45.54,18.74.分析条件:色谱柱beta-dex225手性柱,30m×0.25mm×0.25μm;柱温80℃。保留时间51.6min(s-构型),52.7min(r-构型)。
实施例2
在氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(r)-l2(ar'=ph)(3.0mg)和四氢呋喃(2.0ml),在室温下搅拌至蓝色溶液。给上述体系中加入聚(甲基氢硅氧烷)(180μl)、叔丁醇(140μl),搅拌后加入2-苯基丙烯酸甲酯(162mg),再搅拌反应2h。给反应混合物中加入饱和的nh4cl溶液(4.0ml),继续搅拌20min,分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得到无色的液体2-苯基丙酸甲酯101mg,产率62%;经手性gc分析,ee值为19%,优势构型为(s)-构型。
实施例3
在氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(s)-l3(ar'=ph)(6.1mg)、甲苯(2.0ml),在室温下搅拌至蓝色溶液。给上述液体中加入ph2sih2(184μl)、新戊丁醇(132mg),搅拌5min后加入2-苯基丙烯酸甲酯(152mg),继续搅拌反应2h。给反应混合物中加入饱和nh4cl溶液(4.0ml),继续搅拌20min,分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得到2-苯基丙酸甲酯(111mg,产率72%),gc手性分析,ee22%),优势构型为(s)-构型。
实施例4
在氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(r)-l4(ar'=4-me-3,5-(t-bu)2-ph)(5.6mg)、甲苯(2.0ml),在室温下搅拌至蓝色溶液。降温至-25℃,给上述液体中加入聚(甲基氢硅氧烷)(120μl)、叔丁醇(182μl),搅拌5min后加入2-苯基丙烯酸甲酯(161mg)。搅拌2h后,然后加入饱和nh4cl溶液(4.0ml),继续搅拌30min后分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得产品(149mg,产率91%,ee35%,优势构型为(s)-构型。外消旋体和本反应得到的2-苯基丙酸甲酯分析的手性gc谱图分别见图3、图4。
实施例5
在氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(r)-l4(ar'=4-me-3,5-(t-bu)2-ph)(5.6mg)、二甲苯(2.0ml),在室温下搅拌至蓝色溶液。降温至-30℃,给上述液体中加入聚(甲基氢硅氧烷)(120μl)、甲醇(40μl),搅拌5min后加入2-(4-异丁基苯基)-丙烯酸甲酯(108mg)。搅拌2h后加入饱和nh4cl溶液(4.0ml),继续搅拌30min后分液,水相经用二甲苯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得产品(93mg,产率85%,ee53%,优势构型为(s)-构型。1hnmr(500mhz,cdcl3)δ7.20(d,j=7.9hz,2h),7.10(d,j=7.9hz,2h),3.70(q,j=7.1hz,1h),3.66(s,3h),2.44(d,j=7.2hz,2h),1.87-1.81(m,1h),1.49(d,j=7.2hz,3h),0.90(d,j=6.6hz,6h).13cnmr(126mhz,cdcl3)δ175.39,140.70,137.85,129.49,127.25,52.15,45.16,45.14,30.33,22.54,18.77.外消旋体和本反应得到的2-(4-异丁基苯基)-丙酸甲酯的手性hplc谱图分别见图5、图6。
实施例6
氮气下给氮气置换过的、干燥的休良克瓶中加入cucl(10mg)、kobut(11.2mg)、手性配体l5(ar'=ph)(67mg),thf(2.0ml),搅拌10min后取出其1/10,加入另一反应瓶中,加入thf(2.0ml),降温至-50℃。给上述反应瓶中加入(meo)3sih(127μl)、叔丁醇(92μl),搅拌5min后加入2-(2-氯苯基)丙烯酸甲酯(92mg,0.50mmol)。搅拌1h后加入饱和nh4cl溶液(4.0ml),继续搅拌20min后分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得产品(89mg,产率90%,ee47%)。1hnmr(500mhz,cdcl3)δ7.37(dd,j=7.9,1.4hz,1h),7.31(dd,j=7.7,1.7hz,1h),7.28–7.23(m,1h),7.22–7.17(m,1h),4.22(q,j=7.2hz,1h),3.68(s,3h),1.50(d,j=7.2hz,3h).13cnmr(126mhz,cdcl3)δ174.61,138.57,133.79,129.82,128.53,128.42,127.31,52.31,42.21,17.75。本反应得到的2-(2-氯苯基)丙酸甲酯经手性od-h柱分析的hplc谱图见图7。
实施例7
在氮气置换过的、干燥的反应瓶中加入cu(oac)2·h2o(2.0mg)、(s)-l4(ar'=ph)(5.8mg)、甲苯(2.0ml),室温下搅拌10min。给上述液体中加入ph2sih2(184μl)、叔丁醇(140μl),搅拌5min后加入2-苯基丙烯酸苄酯(232mg),继续搅拌反应2h。之后,给反应混合物中加入饱和nh4cl溶液(4.0ml),继续搅拌20min,分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得到2-苯基丙酸苄酯169mg,产率72%;经hplc分析,ee22%。
实施例8
在氮气置换过的、干燥的休良克瓶中加入cuf(pph3)3·2meoh(4.6mg)和(r)-l4(ar'=4-me-3,5-(t-bu)2-ph)(5.6mg)、甲苯(1.0ml),室温下搅拌10min后取出其中液体0.1ml加入到另一个反应瓶中,降温至-25℃,给后一个反应瓶中加入聚(甲基氢硅氧烷)(120μl)、叔丁醇(182μl),搅拌5min后加入2-(4-甲氧基苯基)丙烯酸甲酯(97mg),搅拌5h。然后给反应瓶中加入饱和nh4cl溶液(4.0ml),继续搅拌30min后分液,水相经用乙酸乙酯(3×5.0ml)萃取,合并的有机相经nacl饱和溶液洗涤、无水硫酸钠干燥、浓缩、柱分离得(4-甲氧基苯基)丙酸甲酯(90mg,产率92%,ee38%)。1hnmr(500mhz,cdcl3)δ7.22(d,j=8.6hz,1h),6.86(d,j=8.7hz,1h),3.78(s,1h),3.72–3.66(q,j=7.2hz,1h),3.65(s,1h),1.47(d,j=7.2hz,3h).13cnmr(126mhz,cdcl3)δ175.39,158.81,132.78,128.58,114.14,55.37,52.09,44.68,18.79。本反应得到的2-(4-甲氧基苯基)丙酸甲酯的手性hplc谱图见图8。
以上所述,仅为本发明创造较佳的具体实施方式,但本发明创造的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明创造披露的技术范围内,根据本发明创造的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明创造的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!