连接酶筛选测定的制作方法
发明领域
本发明涉及用于鉴定mycbp2调节剂的方法。合适的调节剂通过mycbp2泛素e3连接酶活性的调节而鉴定,其中,所述调节经由对两个催化性半胱氨酸(c4520和c4572)中的任一个的共价修饰,或通过阻碍新呈现的动态性的c4520所在的被称为介导环区的运动进行。本发明也涉及含有羟基的小分子及肽作为测定mycbp2连接酶活性的代表底物的用途,和它们在鉴定调节剂的方法中的用途。
背景技术:
利用泛素(ub)和泛素样修饰蛋白(ubls)的蛋白修饰调节着真核生物的大多数方面。ub/ubl缀合是通过由e1活化(e1),e2缀合(e2)和e3连接(e3)酶组成的酶级联实施的。缀合需要初始atp-依赖性硫代酯化步骤和经由在e1s,e2s和e3s中的催化性半胱氨酸的毗邻的多达两个随后的转硫酯化步骤。泛素化通常被认为是赖氨酸残基的翻译后修饰。
与e6-ap羧基末端(hect)同源的e3经历与e2-泛素共价连接的中间体发生的催化性半胱氨酸-依赖性转硫醇反应,从而形成共价连接的e3-泛素中间体。此外,ring-between-ring(rbr)e3则具有规范的连接至辅助域的ring结构域。这包含了催化性半胱氨酸,使得杂合ring/hect机制能够实现。
泛素缀合酶级联被认为涉及广泛的疾病谱。尽管已经鉴别了许多不同的e2和e3酶和种类,但对这些酶中的一些所知甚少。因此,仍然需要分析或进一步分析这些酶的活性。
本发明考虑这些问题而进行了设计。
发明概述
本发明基于由发明人对e3泛素连接酶mycbp2的作用机制的研究。发明人出乎意料地发现,这种连接酶对于例如含有羟基的底物表现出酯化活性。发明人还认为,在e2缀合酶(e2)与在mycbp2内的半胱氨酸残基之间发生转硫醇。这导致泛素的分子内中转,泛素从mycbp2内的半胱氨酸残基到另一个半胱氨酸残基,然后到其底物。
基于作用机制的鉴定可以研究活性mycbp2蛋白与底物的相互作用,无论底物是人造的或内源性的。能够有利地活用这种相互作用,来对于它们对这种相互作用的效果而筛选测试物质。因此,本发明的目的提供基于这种相互作用的筛选测定。本发明的进一步目的在于鉴定和/或提供mycbp2的调节剂,其用于治疗和/或预防,例如与轴突变性相关的损伤和病症的治疗和/或预防。
可以被理解为,除非另有说明,本文所述的任何特征(包括任何随附的权利要求书和图),可以与以下任何方面的任何组合相结合。
根据第一方面,提供用于鉴定mycbp2调节剂的方法,所述方法包括:
a)使mycbp2,其直系同源物,突变体或片段与测试物质接触;
b)提供探针,所述探针能够与活性mycbp2的c4520,c4572和/或介导环区(残基4515-4531)相互作用;和
c)检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段相互作用的水平是否被调节。
为了避免疑义,c4520存在于序列基序gearcdaea内,而c4572存在于序列基序avffcfgtt内。
当靶向任一个经鉴定的半胱氨酸残基时,测试物质通常可以为小分子亲电体,如丙烯酰胺,丙烯腈或丙烯酸酯。由于能够与亲电体反应的硫醇基存在于半胱氨酸残基上,假定mycbp2活性通过以下进行调节:
a)利用亲电子的小分子配体共价修饰c4520;或
b)利用亲电子的小分子配体共价修饰c4572;
或测试小分子可通过破坏介导环区(残基4515-4531)的活动性发挥作用。介导环活动性的破坏可以通过例如,合适的生物物理技术如核磁共振(nmr)光谱,顺磁共振(epr)光谱或荧光共振能量转移(fret)测定。
发明人已经确定,对于含有羟基的小分子及肽,mycbp2具有意料不到的高且混杂的酯化活性。因此,使用小分子羟基化合物作为代表底物(proxysubstrates)可以用于鉴定亲电子的小分子配体。发明人在本文中将这样的代表底物描述为羟基探针。
不意在以理论束缚,发明人认为全长天然mycbp2蛋白的残基c4520和c4572为在e2-泛素和mycbp2之间的转硫醇以及随后的泛素中转所需的,因此这些残基中的一个或多个将理解为包含活性位点。c4572在下游运转并与羟基探针相互作用。在一些实施方式中,活性位点至少包括残基c4520。在这样的实施方式中,可替换探针能够与残基c4520相互作用(如在wo2016/051174中报告的那些)。
在当与不存在所述测试物质时的相互作用的水平相比,所述探针与mycbp2,其直系同源物,突变体或片段相互作用的水平被调节的实施方式中,测试物质将被理解为mycbp2调节剂。
mycbp2为在人类发现的0.5mda的蛋白,其包含c-末端ring结构域。该蛋白在发育的神经系统中的轴突引导和突触形成中起作用。在哺乳动物细胞中,该蛋白调节camp和mtor信号途径,并可能另外调节自噬。在本发明的上下文中,将理解mycbp2,其直系同源物,突变体或片段是指核酸序列或其表达产物,例如dna,cdna,mrna,蛋白或其肽片段。
典型地,mycbp2,其直系同源物,突变体或片段包括蛋白或其肽片段。有利地,这实现在mycbp2,其直系同源物,突变体或片段与探针之间的蛋白-底物相互作用的筛选。
如本文所用,活性mycbp2被理解为指酶促活性mycbp2蛋白。
通过“进行相互作用”或“相互作用”,这应理解为是指所述探针与mycbp2,其直系同源物,突变体或片段的活性位点的缔合。本文描述的探针可以为被泛素分子共价修饰的。
因此,本文使用的术语探针指分子,其能够与活性mycbp2的一个或多个活性位点相互作用。
将理解,如本文所用的mycbp2的残基编号与天然全长mycbp2规范的同种型(uniprot登录号075592:http://www.uniprot.org/uniprot/o75592.)关联,或如在pao等.nature.2018apr;556(7701):381-385中所描述。提及“c”为参考了标准氨基酸编码。因此,“c”指半胱氨酸残基。在本申请的首次提交之后,swissprot对mycbp2的入口已被修改为包括插入序列,所述插入序列在本文讨论的催化性区之外。然而,这造成影响,使序列编号以38个残基的方式改变。然而,发明人仍然保留了如在优先权申请和pao等文献中描述的原始编号,并且技术人员读者通过向其添加38,能够容易地从更新的序列编号特定本文所描述的相关的半胱氨酸残基和环区。
相互作用可包括羟基探针,或可替换探针对mycbp2,其直系同源物,突变体或片段的活性位点的特异性结合。
作为酶,将被理解为存在活性mycbp2的内源性底物。探针可包含mycbp2的天然底物,即已知在体内能够与mycbp2的活性位点相互作用的内源性底物。在一些实施方式中,探针包含活性mycbp2的非内源性底物。通过术语非内源性,将被理解为其包括经修饰的内源性底物,以及尚未已知与mycbp2在体内发生作用的底物(即人造的底物)。
探针可以是经修饰的或未经修饰的。例如,探针可包含经修饰的e2,例如e2-泛素缀合探针。这样的经修饰的探针的例子提供于wo2016/051174中,其细节通过引用并入本文。另外,鉴于我们通过mycbp2证实的新呈现的、混杂的酯化活性,探针分子可以为人造的含有羟基的底物如小分子或肽。如本文所述,活性可以通过测定mycbp2和来自上游e2缀合酶的代表底物依赖性释放的泛素,或代表底物与泛素的直接酯化进行评估。
如本文所用,术语“调节剂”将被理解为相对于正常水平(即,当不存在所述测试物质时的水平),mycbp2调节剂使mycbp2,其直系同源物,突变体或片段的活性发生增加或降低。与不存在所述测试物质时的相互作用的水平相比,相互作用的被调节水平可以作为mycbp2的活性的调节水平的结果。
在一些实施方式中,调节剂通过破坏编码mycbp2,其直系同源物,突变体或片段的核酸序列发挥功能。在一些实施方式中,调节剂通过破坏编码mycbp2,其直系同源物,突变体或片段的活性位点的核酸序列发挥功能。例如,步骤a)可包括使mycbp2,其直系同源物,突变体或片段的核酸序列与测试物质接触,并表达经接触的核酸序列的蛋白或其肽产物。然后能够测量表达的蛋白或肽产物的活性。在这样的实施方式中,步骤c)包括检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段的表达的蛋白或肽产物之间,相互作用的水平是否被调节。在这样的实施方式中,“不存在所述测试物质时”被理解为指其中mycbp2,其直系同源物,突变体或片段的核酸序列在步骤a)中不接触测试物质。
在其他的实施例中,步骤a)可包括使mycbp2,其直系同源物,突变体或片段与测试物质接触,其中,mycbp2,其直系同源物,突变体或片段包括蛋白或其肽片段。在这样的实施方式中,步骤c)包括检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段的蛋白或其肽片段之间,相互作用的水平是否被调节。
调节剂可以为抑制剂,即其降低mycbp2的活性。在这样的实施方式中,步骤c)包括检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段相互作用的水平是否被降低。mycbp2的活性可以被调节剂降低至少10%,至少20%,至少30%,至少40%,至少50%,至少60%,至少70%,至少80%,至少90%或100%。
抑制剂可通过活性位点的共价失活发挥功能。另外,或除此以外,抑制剂可通过破坏探针与mycbp2,其直系同源物,突变体或片段的结合发挥功能。在一些实施方式中,抑制剂通过破坏编码mycbp2,其直系同源物,突变体或片段的核酸序列发挥功能。
破坏核酸序列可包括:核酸序列的切割,核酸序列的部分缺失,核酸序列的降解,核酸序列的不稳定和/或在核酸序列中插入另一核酸序列。
任何合适的物质都可以作为可能的mycbp2调节剂被测试。然而,设想的是,该方法可以使用于鉴定mycbp2调节剂,其用于治疗和/或预防,例如神经元损伤,例如,轴突变性相关的损伤和疾病的治疗和/或预防。在一些实施方式中,测试物质包括sirna,mirna,crispr/cas系统,肽,蛋白,酶,小分子,适体,抗体或抗体片段(如fab或f(ab’)2)片段,scfv抗体或任何其他功能性抗原结合片段。
如本文所用,术语“小分子”为具有不大于2千道尔顿(kda)的分子量的化学化合物。在一些实施方式中,小分子具有不大于1kda,或900道尔顿(da)的分子量。在一些实施方式中,小分子具有不大于700或不大于500da的分子量。小分子可以是有机化合物。示例性化合物可包括丙烯酰胺,丙烯腈和丙烯酸酯。
在一些实施方式中,测试物质包括肽,蛋白,酶,小分子,适体,抗体或抗体片段(如fab或f(ab’)2)片段,scfv抗体或任何其他功能性抗原结合片段。
测试物质可包含适体,抗体或抗体片段(如fab或f(ab’)2)片段,scfv抗体或任何其他功能性抗原结合片段。
在一些实施方式中,测试物质包括crispr/cas系统,sirna或mirna。在这样的实施方式中,步骤a)包括使mycbp2,其直系同源物,突变体或片段的核酸序列与测试物质接触,并表达经接触测试物质的mycbp2,其直系同源物,突变体或片段的蛋白或其肽产物。在这样的实施方式中,步骤c)包括检测与不存在所述测试物质时的相互作用的水平相比,探针与步骤a)的蛋白或肽产物之间,相互作用的水平是否被调节。将被理解为在这样的实施方式中,调节剂通过以下发挥功能:调节mycbp2,其直系同源物,突变体或片段的核酸序列,使得探针与产生的蛋白质或肽产物的相互作用被调节。
本领域技术人员能够了解,crispr/cas系统为一种分子工具,利用crispr/cas系统能够靶向和切割靶核酸序列,通常为dna。靶核酸序列通过向导rna(crisprrna或crrna)被靶向,向导rna与作为反式-活化rna(tracrrna)为人所知的、进一步的小rna形成双链体。双链体与cas蛋白形成复合体,使得cas蛋白作用于靶核酸序列,例如通过切割所选择的核酸序列。
就向导rna对靶核酸序列的靶向而言,需要靶核酸序列下游的“前间区序列邻近基序”(pam)区。
因此,crispr/cas系统被理解为指特异性针对靶核酸序列,cas蛋白和pam区的向导rna。crispr/cas系统将理解为也包含进一步的小rna。在一些实施方式中,crispr/cas系统包括-单向导rna(sgrna),sgrna包含crrna和tracrrna。在一些实施方式中,crispr/cas系统进一步包括修复模板。作为本领域技术人员将理解的,修复模板为核酸序列,其包含针对在切割位点中的插入的特定核酸序列。
在本发明的上下文中,靶核酸序列将被理解为mycbp2,其直系同源物,突变体或片段的核酸序列。靶核酸序列可以为至少20个碱基对。在一些实施方式中,靶核酸序列为不大于70个碱基对。在一些实施方式中,靶核酸序列为至少20个碱基对且不大于55个碱基对。
有利地,因此,crispr/cas系统可以被用来修饰mycbp2,其直系同源物,突变体或片段核酸序列,通过切割和/或特定的核酸的序列插入。然后能够检测经修饰的表达的蛋白或肽的活性。
在测试物质包括crispr/cas系统的实施方式中,可以设想各种方法用于使mycbp2,其直系同源物,突变体或片段靶核酸序列进行接触。例如,使mycbp2,其直系同源物,突变体或片段靶核酸序列进行接触,可包括通过例如,电穿孔,转染或转导将细胞与crispr/cas系统接触。经接触的mycbp2,其直系同源物,突变体或片段靶核酸序列然后能够从细胞分离,或以细胞裂解物形式提供。
用于合并靶核酸序列邻近的pam区,产生特异性向导rna,tracrrna和/或sgrna的合适方法,和使用crispr/cas系统的方法对本领域技术人员而言将会是已知的,并也能容易地从各种参考文献如参考文献42-63获得。
也可以运用如在参考文献64-66中详述的基于网络的工具,来帮助鉴定合适的crispr靶序列。
在一些实施方式中,cas蛋白具有核酸内切酶活性。因此,在这样的实施方式中,cas蛋白能够切割被选择的核酸区。在其他的实施例中,cas蛋白为死亡的核酸酶(即具有降低的或无核酸酶活性)。当cas蛋白为死亡的核酸酶时,cas蛋白可以附着于转录激活剂或转录阻遏物,使得被选择的核酸的表达能够分别被上调或下调。
理想地,cas蛋白包括cas9蛋白。
该方法适用于测试多种测试物质。有利地,这提供了用于快速和精确鉴定mycbp2调节剂的高通量筛选测定。
在一些实施方式中,探针包括羟基,探针能够经由羟基与活性mycbp2相互作用。在探针包含羟基的实施方式中,探针可以能够与残基c4520,任选地与残基c4572相互作用。羟基可以存在于氨基酸中。
相互作用可包括羟基与泛素的酯化,所述酯化通过mycbp2,其直系同源物,突变体或片段进行。
因此,在一些实施方式中,方法进一步包括提供泛素。
探针可包含肽。在一些实施方式中,探针包括小分子。经本文测试并显示为功能性的示例性代表底物为甘油,三(羟甲基)氨基甲烷,hepes(4-(2-羟乙基)-1-哌嗪乙磺酸),丝氨酸,苏氨酸,和小的含有丝氨酸/苏氨酸的肽。然而,任何具有暴露羟基的小分子也许有望充当代表底物。
在探针包含羟基的实施方式中,在探针与mycbp2,其直系同源物,突变体或片段之间,相互作用的水平可以间接地测定,例如通过监测探针分子与泛素的酯化而测定。
大多数已知的e3连接酶呈现对于赖氨酸残基的选择性。由此,许多e3连接酶使底物上的赖氨酸残基泛素化。出乎意料,本发明人发现,mycbp2呈现对于苏氨酸,以及,较小程度的对丝氨酸的选择性,而不是赖氨酸。因此,在一些实施方式中,探针可包含苏氨酸或丝氨酸,优选苏氨酸。
在一些实施方式中,方法进一步包括提供泛素酶级联的一种或多种其他组分。泛素酶级联的其他组分包括atp,e1活化酶,e2缀合酶(也称为e2或e2酶)和/或泛素。通过提供这些组分中的一种或多种,可以全部或部分地重现内源性酶级联。优选,在探针包含羟基的实施方式中,方法进一步包括提供泛素酶级联的其他组分中的一种或多种。在一些实施方式中,方法包括提供atp,e1活化酶,e2和泛素。e2酶可以选自ube2d1,ube2d2,ube2d3,ube2d4和ube2e1的变体。在一些实施方式中,e2酶选自ube2d1,ube2d1c86,ube2d1c86azf3(x)和ube2d3。
在其他的实施例中,探针包含相应于式(i)的活化生物分子
其中,x为生物分子且ewg为吸电子基。在本实施方式中,探针能够通过以下方式与活性mycbp2相互作用,当与活性mycbp2的活性位点的半胱氨酸基团接近时,能够通过点击样(click-like)硫醇加成反应而形成共价键。在这样的实施方式中,步骤c)包括检测可以形成的一种或多种缀合物的形成,其中,与不存在所述测试物质时的相互作用的水平相比,一种或多种缀合物的被调节的形成表示了相互作用的被调节水平。
优选生物分子的含硫部分是为了形成活化生物分子而衍生的。典型的生物分子可以是蛋白或肽,而含硫部分为半胱氨酸,特别是存在于蛋白或肽内的内部半胱氨酸。半胱氨酸可以是在生物分子中天然存在的半胱氨酸,或可以是引入分子内的。
在优选的实施方式中,生物分子为泛素缀合酶(e2)。优选一个或多个催化活性的半胱氨酸残基为衍生的。e2酶可以选自ube2d1,ube2d2,ube2d3,ube2d4和ube2e1。在一些实施方式中,e2酶选自ube2d1,ube2d1c86,ube2d1c86azf3(x)和ube2d3。
在探针包含活化生物分子的实施方式中,探针可以能够与残基c4520相互作用。活化生物分子任选能够与残基c4572相互作用。
在生物分子为e2的实施方式中,在探针与mycbp2,其直系同源物,突变体或片段之间相互作用的水平可以根据泛素从e2的探针依赖性和mycbp2依赖性释放而测定。
吸电子基(ewg)可以为对熟练的技术人员已知的任何合适ewg。合适ewg的例子包括:-no2,-nr3+,-cf3,或其他三卤化物,-cn,-soor,-sooh,-cooh,-coor,-cho,-cor,其中,r通常为h,nh,或c1-c4烷基(例如甲基)或烯基。在优选实施方式中,ewg为-cn,或-co2me。在一个实施方式中,ewg未被进一步取代。在其它实施方式中,ewg可以为了例如提供官能团而被进一步取代,所述官能团能够与再一个分子的一部分发生反应且形成共价键。
例如,ewg基团可以利用包含炔基的分子进行取代。这样的炔基可通过点击化学类型环加成反应与叠氮部分发生反应,而形成1,2,3-三唑。
按照本发明的本实施方式,可以提供活化生物分子缀合探针,其中,活化生物分子缀合探针与式(ii)对应:
其中,x为第一生物分子,ewg为吸电子基且y为另一生物分子。
在一个实施方式中,ewg通过三唑基团的形式结合至生物分子y。按照此实施方式,缀合物(ii),更具体地符合下述缀合物(iii):
这样的缀合物可以按照如下反应形成:
第一生物分子x可以为酶,另一生物分子y可以为例如对于所述酶的底物或配体。例如,所述酶可以为e2泛素缀合酶而底物为泛素。在酶为e2泛素缀合酶而底物为泛素的实施方式中,不需要所述方法提供atp,e1,未经修饰的e2或内源性蛋白底物。
探针可以缀合至表面。探针可以通过在探针和表面之间的被动吸附,例如疏水性或疏水性/离子相互作用而缀合至表面。在一些实施方式中,探针包括用于缀合至表面的部分。例如,探针可包含能够与表面的一部分发生反应而形成共价键的官能团作为所述部分。在这样的实施方式中,探针的官能团可包含胺或巯基。表面部分可包含胺或羧基。在一些实施方式中,探针通过高亲和力非共价反应缀合至表面。
表面可包含一种或多种方便地实现高通量分析的多孔平板。在这样的实施方式中,活性mycbp2,其直系同源物,突变体或片段能够与探针作用,以便活性mycbp2,其直系同源物,突变体或片段被拘束至所述表面。这使得实现例如,通过基于抗体的检测技术如elisa的检测。
本发明的探针可包含标记物,例如亲和标签。如本文所用,术语“标记物”表示生化标志物或标签,即可容易识别的化学部分,例如蛋白,肽,或小分子。标记物可以共价附着至探针。在探针包括n和c末端,例如其中探针包含肽的实施方式中,标记物可以共价附着至n或c末端,优选为n-末端。许多标记物对熟练的技术人员是已知的,包括亲和标记物,例如亲和标签(kimple和sondek,biotechniques(2002),33:578-590),荧光剂(如tamra,dapi,荧光素,cy3,cy5,sybr绿等等),生物素,表位标签或放射性标记物。
在一些实施方式中,方法在不存在内源性mycbp2蛋白底物的情况下实施。方法可以在不存在泛素酶级联的其他成分中的一种或多种,例如atp,e1和/或e2连接酶中的一种或多种的情况下实施。在一些实施方式中,方法不包括提供内源性mycbp2底物,atp,e1和/或e2。有利地,这提供了只需要最少试剂的方法,可以利用最小的成本和复杂性进行实施。
方法可进一步包括提供mycbp2,其直系同源物,突变体或片段的初步步骤。在一些实施方式中,mycbp2,其直系同源物,突变体或片段为内源性的,例如以细胞裂解物形式提供。这提供了方法的生理学环境。例如,在步骤a)包括将突变体的mycbp2与测试物质接触的实施方式中,突变体可已从特定的受试者中分离出来。因此,该方法能够有利地用于鉴定对于特定受试者为特异性的mycbp2调节剂。在调节剂可以用于受试者的治疗和/或预防时,这提供了获得对于受试者的个人化疗法的方法。
在其他的实施例中,mycbp2,其直系同源物,突变体或片段可以为重组的。术语“重组”将被理解为指经遗传改造的,例如包含核酸构建体或从核酸构建体表达的mycbp2,其直系同源物,突变体或片段。
本领域技术人员将因此理解,术语“重组”定义为以核酸构建体形式提供和/或在构建体中表达的内源性mycbp2,其直系同源物,突变体或片段,和/或,以核酸构建体形式提供和/或在核酸构建体中表达的经修饰的mycbp2,其直系同源物,突变体或片段。
在一些实施方式中,在方法中,mycbp2,其直系同源物,突变体或片段可以以过表达形式提供。过表达被理解为指相对于内源性表达水平,表达水平提高。通常,过表达是通过表达核酸构建体而达到的,所述核酸构建体包含mycbp2,其直系同源物,突变体或片段基因或cdna。
在一些实施方式中,步骤a)包括使mycbp2的片段与测试物质接触。对于术语片段,发明人意在包括mycbp2的片段,所述片段能够从天然存在的核苷酸或肽序列变化而来,其条件是所述片段基本上保持mycbp2的生物学活性。所述保持生物学mycbp2的活性,是指相比于天然mycbp2,片段保持至少一部分的酶活性。通常片段保持至少50%,如60%,70%,80%或90%活性。在一些实例中,片段可具有比天然mycbp2更大的酶活性。在一些实施方式中,相比于天然酶,片段可显示在另一个生理学特征上的增加。例如,相比于天然酶,片段可拥有体外和/或在体内的更长的半衰期。
不意在以理论束缚,发明人认为全长天然mycbp2蛋白的残基c4520和c4572是对于在e2-泛素和mycbp2之间的转硫醇和随后的泛素中转所需要的。因此,将理解,片段包含至少mycbp2活性蛋白的活性位点。因此,片段包含残基c4520和c4572中的一个或多个。优选,片段包含至少全长天然mycbp2蛋白的残基c4520~c4572。无意以理论进行束缚,发明人认为由c4520和c4572采用的合作机制的新教导和独特的特性,以及半胱氨酸残基的反应性,使得这些位点特别地享有利用调节剂进行靶向的特征,所述调节剂可具有治疗益处。
除了活性位点以外,优选地,片段具有天然序列的至少5%,至少10%,至少20%,至少30%,至少40%,至少50%,至少60%,至少70%,至少80%或至少90%的序列。
在一些实施方式中,片段进一步包括mycbp2的ring结构域,其被理解为全长天然mycbp2蛋白的残基4390-4441。
在一些实施方式中,相对于全长天然mycbp2蛋白,片段的c-末端是经截短的。
在一些实施方式中,片段包含或由全长天然mycbp2蛋白的残基4390~4572组成。
优选地,片段的n-末端包含或由残基4390组成。在其他的实施例中,片段的n末端包含或由残基4378组成。
在一些实施方式中,片段包含或由残基4378~4572组成。
在一些实施方式中,片段的n末端包含或由全长天然mycbp2蛋白的残基3224组成。在其他的实施例中,片段的n末端包含或由全长天然mycbp2蛋白的残基3156组成。
在一些实施方式中,片段的c-末端包含或由来自全长天然mycbp2蛋白的4572~4640的任何残基组成。
在一些实施方式中,片段包含或由全长天然mycpb2蛋白的残基4390~4640组成。
在一些实施方式中,片段包含或由全长mycbp2蛋白的残基4378-4640组成。该片段包括ring结构域,活性位点和随后的c-末端残基。发明人已发现,该片段将有效地将本发明的羟基探针,例如包含羟基的探针与泛素酯化。
在一些实施方式中,片段包含或由mycbp2的同种型2组成。这应理解为是指全长规范的同种型的mycbp2缺少残基3901~3903。
所述片段可包含直系同源物的片段。
mycbp2的各种直系同源物(orthologues)将会为本领域技术人员已知。例如,mycbp2的直系同源物可包括但不限于,小鼠mycbp2,斑马鱼esrom/phr1,果蝇highwire和秀丽隐杆线虫(c.elegans)rpm-1。
在一些实施方式中,步骤a)包括使mycbp2的直系同源物与测试物质接触。直系同源物可以选自小鼠phr1,斑马鱼esrom/phr1,果蝇highwire和秀丽隐杆线虫rpm-1。在一些实施方式中,直系同源物包含或由果蝇highwire组成。
优选所述方法为体外方法。
对探针与mycbp2,其直系同源物,突变体或片段的相互作用的水平的检测而言,可以为任何合适的分析技术,对本领域技术人员已知的各种技术。
检测可以使用磁性分离,免疫分离,凝胶过滤色谱,亲和色谱,柱色谱,置换色谱,电色谱,气体色谱,高效液相色谱,离子色谱,胶束电动色谱,正相色谱,纸色谱,反相分配色谱,尺寸排阻色谱,薄层色谱,凝胶电泳,离心,粘附,流式细胞仪,或对熟练的技术人员已知的其他技术。
检测可以例如通过还原sds凝胶电泳和免疫印迹,利用对探针或其结合的标签为特异性的抗体而实施。或者,检测可以通过施行质谱法实施。
在实施方式中,其中,相互作用包括与泛素(通过mycbp2,其直系同源物,突变体或片段)进行的探针的羟基的酯化,可以使用允许来自泛素加成的探针的未经修饰的泛素的解析的任何合适技术。例如,羟基可以用适当的报告物标记并检测报告物。报告物可包括但不限于,荧光团,表位标签或生物素。
另外,在相互作用包含探针的羟基与泛素的酯化的实施方式中,“捕获和释放”检测技术可以是合适的,其中,所述酯化利用mycbp2,其直系同源物,突变体或片段进行。不意在以理论束缚,发明人认为在相互作用后,在泛素和羟基之间的酯连接为不稳定的。捕获和释放检测技术可以通过促进酯切割而利用这种不稳定性。例如,酯切割可以由碱如氢氧化物或羟胺介导。经切割的泛素和/或羟基探针可以通过任何合适的分析技术,例如通过elisa或荧光方法如fret或荧光偏振而检测。
在探针包含活化生物分子的实施方式中,检测可以通过任何合适的技术进行,所述技术能够将未经修饰的活化生物分子从结合至mycbp2,其直系同源物,突变体或片段的活化生物分子中解析出来。例如,活化生物分子可包含能通过荧光偏振检测的标记物。在活化生物分子包含e2-泛素缀合探针的实施方式中,检测可以通过使用任何合适的分析技术解析从e2释放的泛素而进行。
检测的其他形式可以包括:活化生物分子,及mycbp2,其直系同源物,突变体或片段可各自包含标记物。一旦两者的标记物紧邻,活化分子与mycbp2,其直系同源物,突变体或片段的结合可以通过fret(荧光共振能量转移)alphascreen(perkinelmer)或htrf(均相时间分辨荧光)技术而检测。
根据第二方面,提供用于鉴定mycbp2调节剂的方法,所述方法包括:
a)使mycbp2,其直系同源物,突变体或片段与测试物质接触;
b)提供能够与活性mycbp2相互作用的探针,所述探针包含羟基,所述羟基能够与mycbp2的c4520并任选与c4572相互作用;和
c)检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段相互作用的水平是否被调节。
本发明也提供了用于鉴定mycbp2调节剂的方法,所述方法包括:
a)使mycbp2,其直系同源物,突变体或片段与测试物质接触;
b)提供探针,所述探针能够与活性mycbp2相互作用;探针包含根据式(i)的活化生物分子,
其中,x为生物分子且ewg为吸电子基;和
c)检测与不存在所述测试物质时的相互作用的水平相比,探针与mycbp2,其直系同源物,突变体或片段相互作用的水平是否被调节。
另一方面,本发明提供试剂盒,所述试剂盒包含如本文定义的探针以及mycbp2,其直系同源物,突变体或片段,用于按照本发明使用。有利地,试剂盒可以使用于分析在来自受试者的样本中分离的mycbp2的活性。分析的活性的水平可以用作受试者的生物标记。生物标记可以使用于确定受试者是否具有特定的疾病或病况。例如,活性的水平高于或低于特定的活性阈值水平可表明受试者具有特定的病症。在一些实例中,活性的水平高于特定的活性阈值水平时,可以表明受试者具有特定的病症,例如神经系统疾病。
按照本发明鉴定的mycbp2调节剂可在治疗或预防神经元损伤的方法中找到用途,其中,mycpb2调节剂能够与mycbp2的活性位点相互作用。
如上所述,发明人认为全长天然mycbp2蛋白的残基c4520和c4572是对于在e2-泛素和mycbp2之间的转硫醇和随后的泛素中转所需要的。因此,活性位点被理解为指残基c4520和/或c4572。
发明人已经观察到mycbp2能够使内源性底物的非赖氨酸的丝氨酸,并优选苏氨酸残基发生泛素化。例如,发明人已经发现mycbp2能够通过酯化使烟酰胺单核苷酸腺苷酸转移酶(nmnat2)泛素化,其为认为参与轴突变性的酶。发明人的观察显示,mycbp2在神经元损伤中的潜在新功能。据此,本发明涵盖mycbp2的调节剂的使用,所述调节剂能够调节mycbp2活性,所述mycbp2活性例如可以从上述鉴定调节剂的方法鉴定。
不意在用任何特定的理论限制,神经元损伤如轴突变性可能至少部分地由于mycbp2的使神经元相关的分子如nmnat2发生泛素化的能力。理想地,因此,调节剂可以是mycbp2的抑制剂。
因此,在一些教导中,调节剂降低mycbp2的活性。mycbp2的活性可以被调节剂降低至少10%,至少20%,至少30%,至少40%,至少50%,至少60%,至少70%,至少80%,至少90%或100%。
如本文所用,“治疗”或“处理”指使关联神经元损伤的症状降低或减轻。如本文所用,“进行预防”或“预防”指保护受试者免受神经元损伤。
调节剂能够例如通过结合与mycbp2的残基c4520和/或c4572相互作用。优选地,通过与这些残基的一个或多个相互作用,mycbp2的活性位点被阻滞,因此mycbp2活性被降低或防止。
神经元损伤可能来自外伤或作为神经毒性疗法,例如化疗的副作用。如本文所用,“外伤”将用于定义神经元损伤,例如作为对受试者的物理力的结果,如跌倒或被对象击中。神经元损伤也可是获得性的,如来自临床使用的神经毒性药物的副作用,所述神经毒性药物如使用在癌症化疗中的那些。
在一些实施方式中,神经元损伤是作为病症的影响,所述病症如神经系统疾病,糖尿病,hiv(人类免疫缺陷病毒)感染,aids(获得性免疫缺陷综合征)和/或缺血。
在一些实施方式中,神经元损伤来自一种或多种神经系统疾病。“神经变性病症”被理解为指神经元在功能和/或结构上退化,和/或死亡的病症。变性通常是渐进的,但在一些实例中可能是突然的。神经变性疾病的实例包括但不限于,阿尔茨海默病,弥漫性路易体病,额颞叶痴呆(fronto-temporaldementia,ftd)(皮克病)(包括ftd亚型行为变体/额叶变体额颞叶痴呆,语义性痴呆(semanticdementia)和进行性非流利性失语(progressivenon-fluentaphasia)),皮质基底节变性(corticobasaldegeneration),嗜银颗粒病(argyrophilicgraindisease),帕金森病,帕金森病痴呆,佩里综合症(perrysyndrome),家族性英国型痴呆(familialbritishdementia),家族性丹麦型痴呆(familialdanishdementia),进行性核上性麻痹(progressivesupranuclearpalsy),多系统萎缩(multiplesystematrophy),路易体病(路易体痴呆),亨廷顿病,脊延髓肌萎缩(kennedy’sdisease,肯尼迪病),齿状核红核苍白球丘脑下部核萎缩(dentatorubral-pallidoluysianatrophy),脊髓小脑性共济失调(spinocerebellarataxias)1,2,3,6,7和17,运动神经元病(肌萎缩侧索硬化),多发性硬化(ms),朊病毒病包括克雅氏病,变异型克雅氏病(牛海绵组织脑病(bovinespongiformencephalopathy)),库鲁病,致命性家族性失眠症(fatalfamilialinsomnia),格-施-沙综合征(gertsmann-straussler-scheinkersyndrome),进行性神经性腓骨肌萎缩(charcot-marietoothdiseases),视神经病变如青光眼,和可变蛋白酶敏感性朊病毒病(variableproteasesensitiveprionopathy)。其他神经变性疾病将是本领域技术人员已知的。
神经变性病症的人体症状可包括:痴呆(包括通过作为本领域技术人员已知的标准测试测量的,记忆力减退和/或认知能力减少)情绪变化,行动不便,言语质量降低或无言语质量,笨拙,平衡困难,不受控制的动作,肢体颤抖,肢体僵硬或运动速度下降。在人体症状的上下文中,“降低”指相对于没有神经变性疾病的个体下降的值。其他的人体症状将是本领域技术人员已知的。
神经变性病症可以选自以下中的一种或多种:阿尔茨海默病,弥漫性路易体病,肌萎缩侧索硬化(als),额颞叶痴呆(ftd)(皮克病)(包括ftd亚型行为变体/额叶变体额颞叶痴呆,语义性痴呆和进行性非流利性失语),皮质基底节变性,嗜银颗粒病,帕金森病,帕金森病痴呆,佩里综合症,家族性英国型痴呆,家族性丹麦型痴呆,进行性核上性麻痹,多系统萎缩,路易体病(路易体痴呆),亨廷顿病,脊延髓肌萎缩(肯尼迪病),齿状核红核苍白球丘脑下部核萎缩,脊髓小脑性共济失调1,2,3,6,7和17,运动神经元病(肌萎缩侧索硬化),多发性硬化(ms),朊病毒病包括克雅氏病,变异型克雅氏病(牛海绵组织脑病),库鲁病,致命性家族性失眠症,格-施-沙综合征,进行性神经性腓骨肌萎缩,视神经病变如青光眼,和可变蛋白酶敏感性朊病毒病。
在一些实施方式中,神经变性病症选自以下中的一种或多种:阿尔茨海默病,帕金森病,肌萎缩侧索硬化(als)亨廷顿病,运动神经元病,格-施-沙综合征,进行性神经性腓骨肌萎缩,视神经病变如青光眼,克雅氏病(prion)和多发性硬化(ms)。
在一些实施方式中,神经变性病症选自以下中的一种或多种:阿尔茨海默病,帕金森病,肌萎缩侧索硬化(als)亨廷顿病,运动神经元病,克雅氏病(prion)和多发性硬化(ms)。
神经变性病症可以选自以下中的一种或多种:阿尔茨海默病,帕金森病,肌萎缩侧索硬化(als)亨廷顿病和运动神经元病。
在一些实施方式中,神经变性病症选自以下中的一种或多种:阿尔茨海默病,帕金森病和亨廷顿病。
神经变性病症可以选自以下中的一种或多种:阿尔茨海默病,帕金森病,肌萎缩侧索硬化(als)和亨廷顿病。在一些实施方式中,神经变性病症选自阿尔茨海默病或帕金森病。
在一些实施方式中,调节剂包括肽,蛋白,酶,适体,抗体或抗体片段(如fab或f(ab’)2)片段,scfv抗体或任何其他功能性抗原结合片段。
在一些实施方式中,调节剂包含或由抗体,抗体片段,肽或适体组成。
调节剂可以为抗体或抗体片段。
在调节剂包含适体的实施方式中,适体可包括dna或rna。
按照本发明鉴定的mycbp2调节剂可在治疗或预防神经元损伤的方法中找到用途,其中,mycpb2调节剂能够破坏mycbp2与其天然底物或配体的结合。
破坏核酸序列可包括:核酸序列的切割,核酸序列的部分缺失,核酸序列的降解,核酸序列的不稳定和/或在核酸序列中插入另一核酸序列。
在一些实施方式中,调节剂包括sirna,mirna或crispr/cas系统。在一些实施方式中,调节剂包括crispr/cas9系统。
在调节剂包括sirna,mirna或如本文所述的crispr/cas系统的实施方式中,治疗或预防神经元损伤的方法可包括:从受试者分离细胞,将细胞与sirna,mirna或crispr/cas系统接触,并施用经接触的细胞至受试者。
在进一步的教导中,教导了治疗或预防受试者中的神经元损伤的方法,所述方法包括施用mycbp2调节剂至受试者。
神经元损伤可以在哺乳动物受试者,例如人中。本发明适用的非人受试者包括:宠物,家养动物,野生动物和牲畜,包括狗,猫,牛,马,绵羊,山羊,鹿和啮齿动物。因此,将理解调节剂可以以治疗有效量提供,所述治疗有效量即,当施用至受试者时足以消除,减少或预防神经元损伤的调节剂的量。
调节剂的施用可以通过任何合适途径进行,包括但不限于,注射(包括静脉内(推注或输注),动脉内,腹膜内,皮下的(推注或输注),心室内,肌内,或蛛网膜下),口服,吸入,局部,经由粘膜(如口服,鼻腔或直肠粘膜),通过喷雾,片剂,经皮贴剂,皮下的种植体形式或栓剂形式递送。施用方式可取决于所处理的神经元损伤。
在调节剂为肽或蛋白的实施方式中,编码肽或蛋白的核酸序列可以在合适的载体,例如质粒,黏粒(cosmid)或病毒载体中提供。因此,也提供了载体(即构建体),其包含编码蛋白或肽的核酸序列。核酸序列优选可操作地连接至合适的启动子。本发明进一步涉及包含所述载体的组合物。
对于为核酸,如sirna,mirnas或crispr/cas系统的调节剂,可以经修饰(例如经由核酸骨架的化学修饰)或在合适的递送系统中递送,所述递送系统保护核酸免受降解和/或免疫系统识别。合适递送系统的例子包括纳米颗粒,脂质颗粒,聚合物介导的递送系统,基于脂质的纳米载体和外泌体。
在一些实施方式中,根据本发明的第五方面,可以施用在0.1μg/kg的体重和1g/kg的体重之间的调节剂的剂量用于神经元损伤的治疗或预防,这取决于使用的具体调节剂。
调节剂可以作为单剂量或作为多剂量施用。多剂量可以在一天内施用(例如2,3或4次剂量,例如以3,6或8小时的间隔)。可酌情地,调节剂可以在定期基础上(例如,每天,每隔一天,或每周)在数天,数周或数月内进行施用。
将被理解,施用的最佳剂量可以由本领域技术人员确定,并将取决于使用的具体调节剂,制剂的强度,施用方式和神经元损伤的进展或严重程度而变化。取决于将被处理的具体受试者的附加因素导致必须调整剂量,所述附加因素包括受试者年龄,重量,性别,饮食,和施用的时间。可以使用已知的步骤,如那些由药品工业(例如在体内测试,临床试验,等)常规使用的那些,来建立用于根据本发明和精确的治疗剂量制度使用的特异性制剂。
现在将通过实施例并参照附图描述本发明的实施方式,其中:
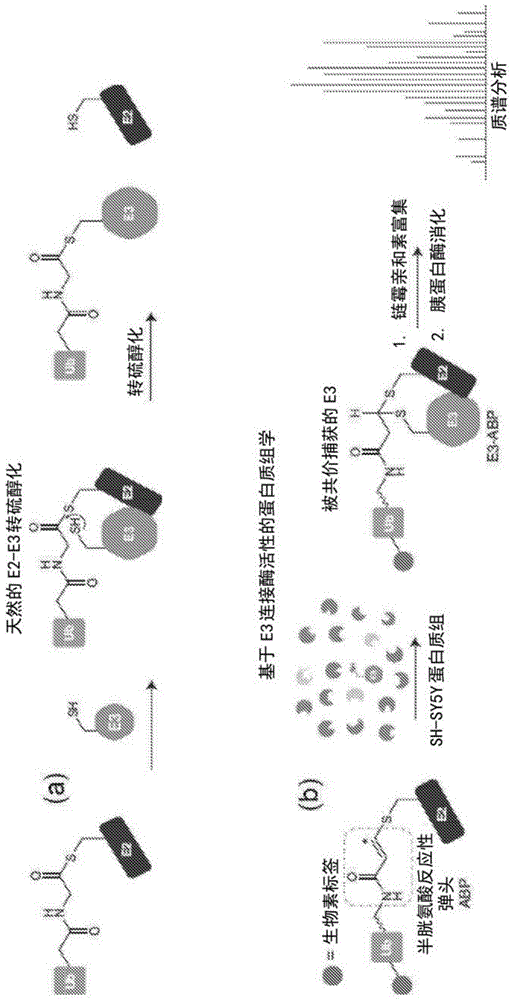
图1显示e3连接酶的基于活性的蛋白质组学;
图2显示生物素化的abp中间体和生物素化的abp的lc-ms表征;
图3显示神经母细胞瘤sh-sy5y细胞的基于活性的蛋白质组学特征分析;
图4显示mycbp2的e3连接酶的新的类型,和半胱氨酸中转机制的数据支持;
图5显示具有高选择性的在mycbp2cat内的abp标记物c4520;
图6显示mycbp2cat的酯化活性,和进一步的支持双半胱氨酸机制的数据,以cis形式运转;
图7显示mycbp2使丝氨酸和苏氨酸泛素化,具有对苏氨酸的选择性;
图8显示mycbp2具有丝氨酸/苏氨酸ub酯化活性,但具有对苏氨酸的偏好;
图9显示mycbp2需要e2;
图10为mycbp2cat晶体模型的结构比较和典型的立体视图;
图11显示mycbp2cat的晶体结构;
图12显示苏氨酸选择性的结构基础,e2-e3中间体的模型和ub中转的模型;
图13显示e2-mycbp2cat复合体的建模;和
图14为对rcre3连接酶机制提出的模型的概要示意。
详细说明
介绍
泛素化通过泛素(ub)从e1活化酶(e1)转移至e2缀合酶(e2)产生共价连接的中间体(e2~ub)而启动1。真正有趣的新基因(reallyinterestingnewgene,ring)类的e3连接酶(e3)经由它们的ring结构域募集e2~ub,并介导ub直接转移至底物2。与e6-ap羧基末端(hect)同源的e3s经历与e2~ub的催化性半胱氨酸依赖性转硫醇反应,而形成共价e3~ub中间体3,4。此外,ring-between-ring(rbr)e3则具有规范的连接至辅助域的ring结构域(环结构域)。这包含了催化性半胱氨酸,使得杂合ring/hect机制能够实现5。泛素化通常被认为是赖氨酸残基的翻译后修饰,作为人e3赋予的非赖氨酸活性仍有待发现。本文中,发明人实现了hect/rbr样e3的基于活性的蛋白特征分析,并揭示了神经元相关的e3mycbp2/phrl作为ring-连接的e3的新的类,其具有酯化活性和对苏氨酸超过丝氨酸的固有选择性。mycbp2包含两种必不可少的催化性半胱氨酸残基,它们经由硫酯中间体将ub中转至底物。这种e3连接酶的新的类,发明人命名为ring-cys-中转(ring-cys-relay,rcr),其晶体表征揭示了对其机制和苏氨酸选择性的洞察。这些发现暗示,通过由e3酶的非赖氨酸泛素化和未被重视的机械学多样性进行的,高等真核生物的细胞调节。
材料和方法
通用材料
所有的dna构建体通过dna测序验证,(medicalresearchcouncilproteinphosphorylationandubiquitylationunit,universityofdundee)。细菌蛋白表达的dna被转化入e.colibl21-de3(merck)。本研究用的生成的所有的cdna质粒和抗体通过发明人的试剂网站(https://mrcppureagents.dundee.ac.uk/)请求是可获得的。除非另有声明,所有的溶剂和试剂购自sigma-aldrich或vwr。
生物素官能化的abp制备
从质粒ptxb1-ubδ74-76-t3c质粒7表达具有gcssgn-末端延伸的ub。也创建了编码ub残基1-74的等效质粒(ptxb1-ubδ75-76-t3c)。如之前描述地获得ub硫酯,所述ub硫酯分别产生半胱氨酸标签化的cys-ub1-73-sr和cys-ub1-74-sr7。延伸的ub1-74是包含其中的,因为这保持了arg74,而arg74与rbre3hoip31形成有利的静电相互作用。cys-ub1-73-sr(30mg)通过添加dmso(116μl),接着是水(456μl)而重构。制备了ez-link碘代乙酰基-peg2-生物素(thermofisher)水性储备液(48mm),并将200μl添加到cys-ub1-73-sr溶液(580μl)中,接着添加900μl除气缓冲液(50mmna2hpo4ph7.5,150mmnacl)。反应在23℃孵育1小时并通过lc-ms监控。蛋白(生物素-ub1-73-sr)然后通过半制备rp-hplc(柱:biobasic-4;部件号:72305-259270)被进一步纯化。在60min内以10mlmin-1的流速应用从20%缓冲液a到50%缓冲液b的梯度(缓冲液a=0.1%tfa在h2o中,缓冲液b=0.1%tfa在乙腈中)。重复上述步骤以生成生物素-ub1-74-sr。收集含有生物素-ub1-7x-sr的hplc级分并冻干(产量:生物素-ub1-73-sr75-85%,生物素-ub1-74-sr40-50%)(图3a和d)。然后如以前描述7制备了生物素-标签化的含有abp的硫代丙烯酰胺弹头,使用e2识别元件ube2d2*,ube2d2*f62a,ube2l3*和ube2l3*f63a提供abp1,3,2和4(图3b,c,e和f)。*表示其中非催化性cys残基被突变为ser的e2。也制备了带有着六组氨酸报告标签及硫代丙烯酰胺弹头的基于ube2d2*和ube2d3*的abp(图5a),分别收获abp5和6。
细胞培养和裂解方案
sh-sy5y细胞如以前描述地培养7。hek293培养(37℃,5%co2)在补充了10%(v/v)胎牛血清(fbs),2.0mml-谷氨酰胺,和抗生素(100单位ml-1青霉素,0.1mgml-1链霉素)的dulbecco′s修饰eagle培养基(dmem)中。细胞转染使用聚乙烯亚胺(polysciences)而根据制造商的说明进行。在裂解两小时前将mg-132(50μm)添加到细胞中。细胞用冰冷的pbs洗涤,并在裂解缓冲液(1%np-40,50mmtris-hclph7.5,1.0mmegta,1.0mmedta,0.27m蔗糖,10mm2-甘油磷酸钠,0.2mm苯甲基磺酰氟(pmsf),1.0mm苯甲脒,1.0mm原钒酸钠,50mm氟化钠和5.0mm焦磷酸钠,50mm碘乙酰胺和completetm,edta-无蛋白酶抑制剂鸡尾酒(roche))中提取。然后将裂解物通过在4℃以14,800rpm离心30min而澄清。收集上清液(总细胞提取物)并通过bradford分析测定蛋白浓度。对于碱不稳定性测试,指定的细胞裂解物被进一步地用0.5m羟胺,ph9.0,在37℃孵育30分钟。
免疫印迹
将样品与nupagelds样本缓冲液(thermofisher)混合,不进行煮沸,并用mops或mes运行缓冲液通过sds-page(4-12%nupage胶,thermofisher)解析,并转到0.45μm硝化纤维素膜(gelifesciences)上。将膜用含有5%(w/v)无脂肪干燥脱脂奶粉(pbs-tm)的pbs-t缓冲液(pbs+0.1%tween-20)在室温封闭1h。随后,膜与指定的抗体在含有5%(w/v)牛血清白蛋白(bsa)的pbs-t中,在4℃探针标记过夜。使用hrp--缀合第二抗体在pbs-tm中在23℃1h而进行检测。使用了ecl蛋白质印迹检测试剂(gelifesciences)用于可视化,可视化按照制造商方案进行。
抗体
经his-标签化的类别用1:10000抗his第一抗体(clontech,#631212)进行探针标记。α微管蛋白(1e4c11)小鼠mab
sh-sy5y细胞的基于活性的蛋白质组学特征分析
sh-sy5y总细胞裂解物(4.5mg,550μl)与abp1,2,3和4(3μm)混合,并在30℃孵育4小时。为了诱导parkin活化细胞,用寡霉素(5μm)和抗霉素a(10μm)(oa)施用3小时。也进行了对照富集,其中不给予探针。将提取物与100μl的piercetmstreptavidinplusultralinktm树脂(thermofisherscientific)混合,并用6%sds溶液(20μl)稀释至终浓度为在磷酸缓冲液中0.2%。样品在4℃孵育4小时,并洗涤(2ml0.2%sds/pbs,2mlpbs,1ml4m脲/pbs,2mlpbs)树脂,然后重悬在190μltris缓冲液(50mmtrisph8,1.5m脲)中。树脂-结合蛋白质用tcep(5mm)在37℃还原30分钟,然后用碘乙酰胺(10mm)在23℃烷基化20分钟。然后添加dtt(10mm),接着用缓冲液(50mmtrisph8,1.5m脲)洗涤至终体积300μl。然后添加胰蛋白酶(2μg),并进一步在37℃孵育14小时。添加三氟乙酸至终浓度0.1%,样品用c18macrospin柱(nestgroupinc)脱盐。lc-ms/ms分析在与ultimatenanoflowhplc系统(dionex)联用的ltqorbitrapvelos设备(thermoscientific)上进行。应用从3%溶剂b运行到99%溶剂b的梯度,345min(溶剂a=0.1%甲酸和3%dmso,在水中;溶剂b=0.08%甲酸和3%dmso,在80%mecn中)。
数据处理
使用mascotserver(matrixscience)针对swissprot数据库和诱饵数据库(decoydatabase)检索了raw文件。应用了具有多达三个缺失的切割的胰蛋白酶特异性。半胱氨酸氨基甲酰化被设定为固定修饰,可变修饰为甲硫氨酸氧化/二氧化。使用了perl脚本来对于每一蛋白从mascot搜索结果中提取等级1的肽的数目,该数字被用作质谱计数的数字。第二perl脚本通过使用e3结构域术语ring,hect,ibr和zf-ubr搜索了人swisspfam_v30数据库而对数据进行过滤。也实施了人工筛选(manualcuration),其包括添加e1酶。具有小于3的质谱计数和相对于对照实验小于14倍的质谱计数富集的任何蛋白质被从列表中省略,在所述对照实验中,不给予abp。然后将成对数据集在prism(graphpad软件)中绘制为柱型图。
mycbp2cat的克隆
从全长addgene质粒#2570扩增了人mycbp2(nm_015057.4)序列。将野生型和突变体片段作为bamhi/not1插入序列亚克隆至pgex6p-1(gelifesciences)中用于细菌表达,或亚克隆至pcdnatm5/frt/to(thermofisher)的含有n-末端myc标签的修饰版本用于哺乳动物表达。
ube1和e2表达和纯化
6his-ube1在sf21细胞中表达,并如以前描述经由其标签纯化32。在整个纯化过程中使用磷酸盐缓冲盐水,并避免含有羟基的化合物。ube2d3在bl21细胞中作为n末端带有6his标签的蛋白(n-terminally6his-taggedprotein)表达,并在ni-nta-琼脂糖中被纯化,最后透析至50mmna2hpo4ph7.5,150mmnacl,0.5mmtcep中。ube2a在e.coli中以gst融合的形式表达,且gst标签以蛋白水解被除去。其余的e2表达为重组细菌蛋白质形式,并利用它们的his标签被纯化,并通过尺寸排阻色谱使用superdex75柱(gelifesciences)进行缓冲液交换,交换至运行缓冲液(50mmna2hpo4ph7.5,150mmnacl,0.5mmtcep,0.015%brij-35)中。
mycbp2和gst-mycbp2的表达和纯化
将gst标签化的mycbp2cat(ser4378-phe4640)的wt和突变体在16℃过夜表达,并使用标准步骤针对谷胱甘肽树脂(expedeon)进行纯化。将gst-标签化的构建体用谷胱甘肽洗脱,并通过用鼻病毒3c蛋白酶的树脂上的切割而获得脱标记的构建体。将蛋白质缓冲液交换至50mmtris-hclph7.5,150mmnacl,1.0mmtcep缓冲液中,并闪冻而储存于-80℃。
nmnat2的表达和纯化
nmnat2在bl21(de3)细胞中表达为具有6his-sumo标签,用0.1mmiptg诱导并在16℃孵育进行表达。收集细胞并使用标准方案在缓冲液(50mmtris-hcl(ph7.5),250mmnacl,0.2mmegta,20mm咪唑,20mml-精氨酸,0.015%brij35,1mm亮肽素,1mmpefabloc,1mmdtt中裂解,蛋白在ni-nta-琼脂糖中纯化。洗脱的蛋白在针对pbs,20mml-精氨酸,1mmdtt的透析期间用his-senp1蛋白酶孵育。标签和蛋白酶针对ni-nta-琼脂糖耗尽,nmnat2被浓缩并在superdex75hr10/30上进行层析而进入缓冲液(pbs,20mml-精氨酸)。
mycbp2半胱氨酸突变体的基于活性的蛋白特征分析
指定的mycbp2突变体被稀释在tris缓冲液(50mmtris-hclph7.5,150mmnacl)中,使终浓度为3μm。添加探针6(12μm)并用e3连接酶在30℃孵育四小时。通过添加4xlds上样缓冲液(补充~680mm2-巯基乙醇)而将反应淬灭,样品通过sds-page(4-12%nupage胶)解析,接着进行考马斯染色或抗his免疫印迹。
探针标记的mycbp2的trypticms/ms测序
使用abp6的交联ms如以前描述7地实施。总结而言,与abp-标记的wtmycbp2相应的考马斯染色sds-page条带通过lc-ms/ms,使用与ultimatenanoflowhplc系统(dionex)联用的orbitrapfusiontmtribridtm质谱仪(thermoscientific)进行分析。应用从0%溶剂a运行到60%溶剂b的梯度,120min(溶剂a=0.1%甲酸,在水中;溶剂b=0.08%甲酸,在80%mecn中)。通过hcd生成片段离子,并且将1+,2+和3+前体离子排除。使用plinksoftware33针对ube2d3*和mycbp2序列检索了raw数据,利用了胰蛋白酶特异性(多达2个缺失的切割)。ms/ms片段离子质量值的误差窗口被设置为软件默认的20ppm。手动添加了306.1805da的交联剂单一同位素的质量,其解释了与双硫醚的形成相关联的理论上的质量差异,所述双硫醚在衍生自探针6的2个cys残基之间形成,这基于ube2d3*并含有硫代丙烯酰胺avs弹头7。
tris/甘油介导的e2释放分析
分析在含有指定的mycbp2突变体(15μm),ube1(1.5μm),ube2d3(15μm),ub(37μm)和atp(10mm)的缓冲液(50mmtris-hclph7.5,150mmnacl,0.5mmtcep,5mmmgcl2)中实施。反应在37℃孵育30分钟。反应通过添加4xlds上样缓冲液(具有或没有~680mm2-巯基乙醇)而终止。将c4572s样本进一步用0.14nnaoh在37℃孵育20分钟,并将样品通过sds-page(4-12%nupage胶)解析,并通过考马斯染色可视化。
亲核试剂释放分析的lc-ms分析
反应如在释放分析中描述的那样制备。在30分钟后,反应使用agilent1200/6130lc-ms系统(agilenttechnologies),使用10-75%梯度,20分钟(缓冲液a=水+0.05%tfa,缓冲液b=乙腈+0.04%tfa)而进行分析。
cy3b-ub的制备
在细菌中从pet质粒(ronaldhay友情提供,universityofdundee)表达了ub,所述ub带有tev蛋白酶可切割的n-末端六组氨酸标签接着是acg基序。将蛋白通过ni亲和色谱法纯化,用tev蛋白酶从标签上切割下来,然后进行缓冲液交换至反应缓冲液(50mmhepes,ph7.5,0.5mmtcep)中。蛋白被浓缩至2mgml-1,并将221μl(50nmol)与cy3b-马来酰亚胺(150nmol,gelifesciences)混合,使得终体积为300μl,并在25℃搅动2h。然后,经标记的蛋白进一步地用p2centri-pure脱盐柱(empbiotech)利用脱气的缓冲液(50mmna2hpo4,150mmnacl)纯化。
mycbp2硫酯/酯捕获分析
ube1(2μm)与cy3b-ub(1μm)在缓冲液(40mmna2hpo4-hclph7.5,150mmnacl,0.5mmtcep,5mmmgcl2)中混合(图2e,泳道1,2)。然后,通过添加atp(5mm)并在25℃孵育10分钟而启动反应。取样品(泳道3,4)并与ube2d3(10μm)结合。在25℃进一步10分钟,之后取样品(泳道5,6)并与gst-mycbp2cat(wt,c4520s,c4520a,c4572s,c4572a,c4520a/c4572s或c4520s/c4572a)(15μm)组合。反应在25℃孵育30秒,并通过添加4xlds上样缓冲液(非还原的或还原的)而终止。对于ub~gst-mycbp2catc4572s酯键切割,在e3与e1/e2混合物反应30秒后添加0.14nnaoh,然后在37℃进一步孵育20分钟。然后用chemidoc凝胶成像系统(biorad)扫描凝胶。
多周转氨基酸和肽组释放分析
将氨基酸的储备液(0.5m)溶解在mq水中,并将ph调整至ph~8。序列ac-egxgn-nh2(x=k,s或t)的肽获自bio-synthesisinc.将储备肽溶液(200mm)溶解在mq水中,并将ph调整至ph~8。ane2(ube2d3)负载反应在含有ube1(250-500nm),ube2d3(20μm),ub(50μm),或cy3b-ub(25μm),mgcl2(5mm)和atp10(mm)的缓冲液(40mmna2hpo4-hclph8.0,150mmnacl,0.5mmtcep)中实施。反应在37℃孵育15分钟,然后在23℃平衡3分钟。然后添加等体积的含有小分子/肽亲核试剂(100mm)的亲核试剂样本和gst-mycbp2(10μm),并在23℃孵育。在所规定的时间点取样品,并如tris/甘油介导的e2释放分析中描述地进行分析。
使用chemidoc凝胶成像系统(biorad)将cy3b-ub可视化。lc-ms如tris/甘油释放中描述地实施,但氨基酸底物样品通过添加2:1份猝灭溶液(75%乙腈,2%tfa)猝灭,而肽底物样品通过添加1∶1份猝灭溶液猝灭。
多周转e2释放组
如氨基酸组中描述地,对e2利用gst-mycbp2cat筛选了苏氨酸释放活性。也将e2在苏氨酸存在,但gst-mycbp2cat不存在的情况下孵育。这些样品提供了参考,以区分固有的e2~ub不稳定性与e3依赖性释放。
利用凝胶中荧光的单周转e2突变体释放
利用cy3b标记的ub(12.5μm)向e2突变体16,17,34-36(10μm)负载,使得终体积为12μl,在37℃20分钟,然后在23℃冷却3分钟。然后通过添加作为mln4924衍生物的化合物1(25μm)37来阻滞e2的再负载,所述化合物1抑制e1,然后进一步孵育15分钟。然后将混合物与12μl的gst-mycbp2cat(5μm)和苏氨酸(100mm)混合,并在23℃孵育所规定的时间。所述分析如多周转分析中地实施。为了解释固有e2~ub不稳定性,针对不给予e3的平行孵育计算了均值%释放(n=2)。数据使用prism(graphpad)绘图。
arih1和ube3c的表达和纯化
arih1残基1-394(dundeeclonedu24260)以n-末端带有gst-标签的融合蛋白形式在bl21细胞中表达。ube3c残基641-1083(dundeeclonedu45301)以n-末端带有gst-标签的融合蛋白形式,在sf21细胞中使用杆状病毒感染系统表达。
对e3-底物依赖性单周转e2~ub释放计算了观察到的速率常数
利用cy3b标记的ub(8μm)向ube2d3或ube2l3(5μm)负载,使得终体积为30μl,在37℃25分钟,然后在23℃孵育3分钟。对于e2~ub释放的单周转条件通过利用作为mln4924衍生物的化合物1(25μm)的e1抑制达到,然后进一步孵育15分钟。混合物然后与30μl的mycbp2cat或arih11-394(hhari)或ube3c641-1083(1μm)和苏氨酸(100mm)混合,并在23℃孵育所规定的时间。样品用非还原4xlds上样缓冲液猝灭,并通过sds-page(bis-tris4-12%)解析。然后用chemidoc凝胶成像系统(biorad)扫描凝胶,随后进行考马斯染色。e2~ub信号使用fiji软件定量。使用prism(graphpad软件)将反应进展曲线拟合为单指数函数,由此获得观察到的速率常数。
mycbp2结晶
mycbp2如未标签化的蛋白中描述地表达。在标签的蛋白酶切割后,蛋白通过尺寸排阻色谱使用
利用多角度光散射(sec-mals)的尺寸排阻色谱
sec-mals测试在ultimate3000hplc系统(dionex)上进行,所述系统具有线内minidawntreosmals检测器和optilabt-rex折射率检测器(wyatt)。而且,蛋白的洗脱曲线也通过在280nm的uv吸光度监控。使用了superdex7510/300gl柱(gelifesciences)。应用了缓冲液条件50mmna2hpo4ph7.5,nacl150mm,1.0mmtcep和流速0.3mlmin-1。用dionex自动进样器将样本(50μl,5.5mgml-1)加样到柱上。使用astra软件v6.0.0.108(wyatt)计算了摩尔质量生成的洗脱峰。
介导环建模
建立了介导环残基并在bioluminate软件
nmnat2泛素化分析
将nmnat2(5μm)与e1(500nm),ube2d3(10μm),mycbp2cat(10μm),ub(50μm),atp(10mm)混合,并与10xph7.5缓冲液(40mmna2h2po4ph7.5,150mmnacl,5mmmgcl2,0.5mmtcep)制成。反应在37℃孵育1小时,并通过添加4xlds上样缓冲液(非还原的或还原的)而终止。对于碱不稳定性测试,向反应补充了0.14nnaoh,然后在37℃进一步孵育20分钟。
生物信息学分析
属于rcr家族的蛋白质通过广义特征搜索(generalizedprofilesearch)鉴定。总共鉴别了671个这样的序列。序列使用pftools软件包,通过特征引导的比对(profile-guidedalignment)进行对齐。为了从各种分类群中鉴定出典型的序列,使用了belvu程序(sangerinstitute)来除去与其他序列具有>80%同一性的序列。手动除去了截短的和错误组装的蛋白质,得到130个典型的tc结构域序列。
数据可用性声明
坐标已存入蛋白数据库(pdbid5o6c)。
结果
发明人制备了发明人最近开发的基于活性的探针(abp)6的生物素化的变体,其特征在于hect/rbre3的标志性的转硫醇活性(图1a和图2)7。将abp技术与质谱法连接,使得实现在神经母细胞瘤sh-sy5y细胞提取物中e3活性的并行特征分析8,9(图1b)。e3使用标准进行了过滤,所述标准确保所检测的e3的至少一个子集的信号与e3活性和/或丰度相关(图3)。达到了对~50个已知的hect/rbre3的~80%的特征分析,但意料不到的是,缺乏hect或rbr辅助域的33ringe3也发生了富集(图1c-e)。为了探索迄今为止尚未发现的ring-连接e3被标记的可能性,发明人专注于mycbp2/phr1(myc-结合蛋白2;pam/highwire/rpm-1)(图1c)。mycbp2是一个大的0.5mda的神经元相关的蛋白,其包含c-末端ring结构域(图4a),并参与包括神经系统发展和轴突变性的调节的细胞过程的范围10-12。
mycbp2的重组c-末端版包含ring结构域(残基4378-4640;mycbp2cat;图4a)和未被描述的c-末端半胱氨酸-丰富区,所述重组c-末端版经历强大的abp标记,具有与已知表明了转硫醇活性的e3同等的效率7,13(图5a)。为了映射推定的催化性半胱氨酸,发明人使用了基于abp的特征分析与abp-交联ms的组合7(图2b,图5b,c,)。数据支持c4520为推定的催化性残基。发明人接下来测定了野生型(wt)e3活性,但未能检测到自动泛素化或自由ub链形成。然而,发明人观察到ub从e2~ub的快速e3-依赖性释放,暗示未知小分子亲核受体的存在(图2c)。液相色谱-质谱法(lc-ms)分析显示,ub被定量地转化成具有相应于与tris(羟甲基)氨基甲烷(tris)和甘油的缩合产物的质量的两种(8639和8668da),这两者分别以常规工作浓度50mm和~65mm存在于发明人的分析缓冲液中。由于在这些亲核试剂内的常见羟基功能性,mycbp2似乎具有酯化活性(图9a,b)。发现活性依赖于c4520,这与它形成硫酯连接的e3~ub中间体一致4,5(图4c)。
意料不到的是,mycbp2c4572s突变体保持了活性,但形成了离散的单ub加合物,所述加合物对硫解是抗性的,但在碱处理后可逆(图4c,d和图6c)5。这可能是发生了突变的s4572残基有助于经由不太短暂的(氧基)酯-连接中间体的形成进行催化,所述中间体在ub和s4572之间形成,保持了修饰底物的能力。发明人假设c4520和c4572两者都是催化性残基,所述催化性残基以串联方式,通过将ub从一个半胱氨酸中转到其他而发挥功能,所述中转通过分子内转硫醇反应进行。为了测试这种中转机制,发明人进行了基于凝胶的硫酯/酯捕获分析14(图4e和图6d),并在wtmycbp2cat上观察到硫醇敏感性ub加合物,而用c4520s突变体则没有观察到(图4e)。与早期测试一致地(图4c),经历加合物形成的c4572s为硫醇耐性,但碱不稳定性(图4e)。因此,如果在ub和c4520之间形成了瞬时的硫酯中间体,则不起反应的c4572a突变体应该将其稳定。事实上,相对于野生型中,在c4572a突变体上的硫醇敏感性加合物形成发生提高,并据推测是经由硫酯键连接的(图4e)。用c4520a/c4572s双突变体没有检测到加合物形成,这支持了加合物形成是连接至c4520残基的(图4e)。在不存在直接证据证明cys-至-cysub转移的情况下,发明人不能正式排除其他可能性。然而,现有的数据与在中转机制中发挥功能的必不可少的半胱氨酸一致。mycbp2cat的突变分析和尺寸排阻色谱-多角度光光散射(sec-mals)(图6e,f)提示,提议的中转机制需要在相同分子中的两种必不可少的半胱氨酸,这与分子内中转机制一致。
鉴于观察到的酯化活性,发明人尝试鉴定mycbp2的氨基酸底物,所述鉴定通过筛选一组氨基酸进行5,其中向苏氨酸的释放活性惊人地提高。产物形成依赖于c4520(图7a,b和图8a-d)。基于ms的定量表明,与丝氨酸相比,对于苏氨酸为~10倍的选择性(图8e)。尽管观察到低水平的赖氨酸修饰,但这独立于mycbp2cat5。在肽背景中也保持了苏氨酸选择性(3倍)(图7c和图8f-h)。此外,在mycbp2cat的存在情况下赖氨酸肽的基础泛素化部分被抑制,强调其缺乏赖氨酸活性(图7c)。综上所述,发明人的测试显示,mycbp2为e3酶的新的类,其经由两种必不可少的半胱氨酸运转,促进ub对羟基的修饰,且与丝氨酸相比,使苏氨酸与ub选择性更高地酯化。由于mycbp2使用了一种新机制,发明人将其称为ring-cys-中转(rcr)e3。发明人标准化了mycbp2苏氨酸酯化活性的催化性效率,发现所述效率介于充分表征的hect和rbre3赖氨酸氨解活性之间5,15(图8i-k)。e2突变分析5,16-19进一步支持了mycbp2cat为e3的新的类(图9a,b和方法)。为了查明功能性的e2伙伴而测试了17种e2,但只有ube2d1,ube2d3和ube2e1证实了强大的活性(图9c)。
mycbp2通过不稳定的烟酰胺单核苷酸腺苷酸转移酶(nmnat2)促进wallerian轴突变性20。发明人接下来测试了在体外,mycbp2cat是否能够通过酯化使nmnat2泛素化(图7d)。尽管含有13个赖氨酸残基,nmnat2经历了氢氧化物不稳定的但硫醇耐性的泛素化,表明mycbp2能够靶向其推定的底物之一中的羟基残基20。细胞底物识别是通过skp1/fbox45底物受体共复合体介导的,skp1/fbox45底物受体共复合体结合至mycbp2cat区n-末端~1940个残基的位点(图7a)21。nmnat2也经历了棕榈酰化和轴突运输22,使得其泛素化的重建(reconstitution)和细胞研究极其具有挑战。然而,为了建立mycbp2cat是否保持了在细胞中的非赖氨酸活性,发明人注视了其在瞬时转染入人胚胎肾293细胞后的自动泛素化。观察到碱不稳定的(但硫醇耐性)的泛素化,且依赖于c4520(图7e)。这表明mycbp2能够保持在细胞中对羟基氨基酸的特异性,且这活性仍然依赖于上游催化性残基,发明人暗示具有ub中转机制。
为了进一步验证ring-cys-中转模型和丝氨酸/苏氨酸活性,发明人使mycbp2cat(残基4378-4640)结晶并解析晶体结构至分辨率
晶体封装显示,在n-末端内的对称性相关mycbp2cat分子(t4380sym)t4380,被放置在邻近酯化位点,在那里它形成了许多底物样相互作用(图12a和b)。首先,t4380sym的β-羟基形成e4534和h4583的补充,并形成潜在的三元组(图12a)。因此,t4380sym的β-氧原子似乎对于去质子化和亲核攻击是首要的。当硫酯连接至c4572,一个催化性的、生产性的亲电子的中心为ub的c-末端。即使在本发明的结构中不存在这ub分子,c4572硫原子距离t4380sym的β-氧原子为3.8a。因此,该结构似乎是准确反映了催化性中间体,该催化性中间体准备好了通过其β-羟基的酯化来经历苏氨酸泛素化(图12a)。此外,phe残基的子集群(f4573,f4578和f4586),其与t4380sym的β-甲基基团邻近,(图12a和b)形成了t4380symβ-甲基基团对接进入的、充分定义的疏水性口袋,并似乎是对于苏氨酸侧链的积极的选择性决定因素。这些残基的提议的功能在苏氨酸释放分析中验证(图12c)。h4583n突变取消了与作为通用基底发挥作用相一致的活性。根据预期的缺陷,h4583n突变体也经历了提高的,硫醇敏感性的,ub加合物的形成,所述缺陷在呈递对于c4572硫酯反应性的底物亲核试剂中(图13a)。苯丙氨酸簇的保守摄动也显著地降低了苏氨酸释放活性(图5c)。e4534的摄动没有减少活性,因此其精确的作用仍然不清楚。
ring结构域与e216-18,23结合的保守允许了e2-rcre3连接酶复合体的建模,其几何上与在e2~ub和mycbp2catc4520之间的转硫醇相容(图5d和图13b)。为了模拟随后的ub中转至c4572需要的构象,发明人用连接至c4520的glygly二肽硫酯建模了缺少介导环的残基,如果本发明的中转模型有效,则ubc-末端的代表将会经由与e2~ub的转硫醇而被转入(图13c-e)。为了支持这种机制,ub硫酯的羰基c原子可以定位在c4572巯基硫原子附近
尽管对非赖氨酸泛素化已有报道25-27,但对优先进行该功能的人e3连接酶仍然难以捉摸。发明人表征的在mycbp2中发现的新型rcre3连接酶,提示通过酯化的泛素化对高等真核生物是固有的,并可能是突触发展和轴突变性的调节剂。此外,虽然没有报告非蛋白ub底物(例如脂质,碳水化合物),但考虑到mycbp2对于小分子羟基化合物的高酯化活性,这仍然是可能的。目前尚不清楚为什么会发展出提议的中转(图14a)机制。然而,转硫醇为不依赖辅因子的过程,提供了ub穿梭整个泛素系统的简便手段1。发明人推测,在空间上,利用与结构上刚性和高保守的e2泛素缀合结构域(ubc)28的直接e2-e3转硫醇,在酯化位点与丝氨酸/苏氨酸活性是互斥的,而介导环的进化解决了这种相容性问题。生物信息学分析显示,在几乎所有的动物中发现了mycbp2直系同源物,但人同源物不太可能存在(表2)。
讨论
通过mycbp2抑制进行稳定nmnat2是有希望的治疗策略,用于减轻在损伤和施用化疗后的神经元损伤10,29,30,并减缓在包括阿尔茨海默和帕金森在内的一系列神经变性疾病的进展29。这种明显的ub中转机制和对导致其的分子机构的结构表征开辟了用于治疗一系列神经病症的新的医学可能。
参考文献
1hershko.a.&ciechanover,a.theubiquitinsystem.annurevbiochem67,425-479,(1998).
2deshaies,r.j.&joazeiro,c.a.ringdomaine3ubiquitinligases.annurevbiochem78.399-434,(2009).
3huibregtse,j.m.,scheffner,m.,beaudenon,s.&howley,p.m.afamilyofproteinsstructurallyandfunctionallyrelatedtothee6-apubiquitin-proteinligase.procnatlacadsciusa92,2563-2567(1995).
4scheffner,m.,nuber,u.&huibregtse,j.m.proteinubiquitinationinvolvingane1-e2-e3enzymeubiquitinthioestercascade.nature373,81-83,(1995).
5wenzel,d.m.,lissounov,a.,brzovic,p..s.&klevit,r.e.ubch7reactivityprofilerevealsparkinandhharitobering/hecthybrids.nature474,105-108,(2011).
6hewings,d.s.,flygare,j.a.,bogyo,m.&wertz,i.e.activity-basedprobesfortheubiquitinconjugation-deconjugationmachinery:newchemistries,newtools,andnewinsights.febsj284,1555-1576,(2017).
7pao,k.c.etal.probesofubiquitine3ligasesenablesystematicdissectionofparkinactivation.natchembiol12,324-331,(2016).
8niphakis,m.j.&cravatt,b.f.enzymeinhibitordiscoverybyactivity-basedproteinprofiling.annurevbiochem83,341-377,(2014).
9borodovsky,a.etal.chemistry-basedfunctionalproteomicsrevealsnovelmembersofthedeubiquitinatingenzymefamily.chembiol9,1149-1159(2002).
10zhen,m,huang,x.,bamber,b.&jin,y.regulationofpresynapticterminalorganizationbyc.elegansrpm-1,aputativeguaninenucleotideexchangerwitharing-h2fingerdomain.neuron26,331-343(2000).
11wan,h.i.etal.highwireregulatessynapticgrowthindrosophila.neuron26,313-329(2000).
12grill,b.,murphey,r.k.&borgen,m.a.thephrproteins:intracellularsignalinghubsinneuronaldevelopmentandaxondegeneration.neuraldev11,8(2016).
13byrne,r.,mund,t.&licchesi,j.activity-basedprobesforhecte3ubiquitinligases.chembiochem,18,1415-1427(2017).
14stieglitz,b.,morris-davies,a.c.,koliopoulos,m.g.,christodoulou,e.&rittinger,k.lubacsynthesizeslinearubiquitinchainsviaathioesterintermediate.emborep13,840-846,(2012).
15you,j.&pickart,c.m.ahectdomaine3enzymeassemblesnovelpolyubiquitinchains.jbiolchem276,19871-19878(2001).
16plechanovova,a.,jaffray,e.g.,tatham,m.h.,naismith,j.h.&hay,r.t.structureofaringe3ligaseandubiquitin-loadede2primedforcatalysis.nature489,115-120,(2012).
17dou,h.,buetow,l.,sibbet,g.j.,cameron,k.&huang,d.t.birc7-e2ubiquitinconjugatestructurerevealsthemechanismofubiquitintransferbyaringdimer.natstructmolbiol19,876-883,(2012).
18lechtenberg,b.c.etal.structureofahoip/e2~ubiquitincomplexrevealsrbre3ligasemechanismandregulation.nature529,546-550,(2016).
19dove,k.k.etal.structuralstudiesofhhari/ubch7~ubrevealuniquee2~ubconformationalrestrictionbyrbrring1.structure25,890-900e895,(2017).
20xiong,x.etal.thehighwireubiquitinligasepromotesaxonaldegenerationbytuninglevelsofnmnatprotein.plosbiol10,e1001440,(2012).
21babetto,e.,beirowski,b.,russler,e.v.,milbrandt,j.&diantonio,a.thephr1ubiquitinligasepromotesinjury-inducedaxonself-destruction.cellrep3,1422-1429,(2013).
22milde,s.,gilley,j.&coleman,m.p.subcellularlocalizationdeterminesthestabilityandaxonprotectivecapacityofaxonsurvivalfactornmnat2.plosbiol11,e1001539,(2013).
23zheng,n.,wang,p.,jeffrey,p.d.&pavletich,n.p.structureofac-cbl-ubch7complex:ringdomainfunetioninubiquitin-proteinligases.cell102,533-539(2000).
24olsen,s.k.&lima,c.d.structureofaubiquitine1-e2complex:insightstoe1-e2thioestertransfer.molcell49,884-896,(2013).
25cadwell,k.&coscoy,l.ubiquitinationonnonlysineresiduesbyavirale3ubiquitinligase.science309,127-130,(2005).
26shimizu,y.,okuda-shimizu,y.&hendershot,l.m.ubiquitylationofaneradsubstrateoccursonmultipletypesofaminoacids.molcell40,917-926,(2010).
27wang,x.,herr,r.a.&hansen,t.h.ubiquitinationofsubstratesbyesterification.traffic13,19-24,(2012).
28alpi,a.f.,chaugule,v.&walden,h.mechanismanddiseaseassociationofe2-conjugatingenzymes:lessonsfromube2tandube2l3.biochemj473,3401-3419,(2016).
29conforti,l.,gilley,j.&coleman,m.p.walleriandegeneration:anemergingaxondeathpathwaylinkinginjuryanddisease.natrevneurosci15,394-409,(2014).
30ali,y.o.,bradley,g.&lu,h.c.screeningwithannmnat2-msdplatformidentifiessmallmoleculesthatmodulatenmnat2levelsincorticalneurons.scirep7,43846,(2017).
31stieglitz,b.etal.structuralbasisforligase-specificconjugationoflinearubiquitinchainsbyhoip.nature503,422-426,(2013).
32stanley,m.etal.orthogonalthiolfunctionalizationatasingleatomiccenterforprofilingtransthiolationactivityofe1activatingenzymes.acschembiol10,1542-1554,(2015).
33yang,b.etal.identificationofcross-linkedpeptidesfromcomplexsamples.natmethods9,904-906,(2012).
34pruneda,j.n.etal.structureofane3:e2~ubcomplexrevealsanallostericmechanismsharedamongring/u-boxligases.molcell47,933-942,(2012).
35wu,p.y.etal.aconservedcatalyticresidueintheubiquitin-conjugatingenzymefamily.emboj22,5241-5250,(2003)
36yunus,a.a.&lima,c.d.lysineactivationandfunctionalanalysisofe2-mediatedconjugationinthesumopathway.natstructmolbiol13,491-499,(2006).
37brownell,j.e.etal.substrate-assistedinhibitionofubiquitin-likeprotein-activatingenzymes:thenedd8e1inhibitormln4924formsanedd8-ampmimeticinsitu.molcell37,102-111,(2010).
38langer,g.,cohen,s.x.,lamzin,v.s.&perrakis,a.automatedmacromolecularmodelbuildingforx-raycrystallographyusingarp/warpversion7.natprotoc3,1171-1179,(2008).
39emsley,p.,lohkamp,b.,scott,w.g.&cowtan,k.featuresanddevelopmentofcoot.actacrystallogrdbiolcrystallogr66,486-501,(2010).
40murshudov,g.n.etal.refmac5fortherefinementofmacromolecularcrystalstructures.actacrystallogrdbiolcrystallogr67,355-367,(2011).
41lovell,s.c.etal.structurevalidationbycαgeometry:phi,psiandcβdeviation.proteins50,437-450,(2003).
42jinek,m.,etal.(2012)science,337,816-821.
43overballe-petersen,s.,etal.(2013)proc.natl.acad.sci.u.s.a.110,19860-19865.
44gong,c.,etal.(2005)nat.struct.mol.biol.12,304-312.
45davis,l.,maizels,n.(2014)proc.natl.acad.sci.usa,111,e924-932.
46ran,f.a.,etal.(2013)cell,154,1380-1389.
47qi,l.s.,etal.(2013)cell,152,1173-1183.
48gasiunas,g.,etal.(2012)proc.natl.acad.sci.usa,109,e2579-2586.
49maede,m.l.,etal.(2013)nat.methods,10,977-979
50gilbert,l.a.,etal.(2013)cell,154,442-451.
51hu,j.,etal.(2014)nucleicacidsres.doi:10.1093/nar/gku109.
52perez-pinera,p.,etal.(2013)nat.methods,10,239-242.
53chen,b.,etal.(2013)cell,155,1479-1491.
54seung,w.,etal.(2014)genomeres.24,132-141.
55miller,j.c.,etal.(2011).nat.biotechnol.29,143-148.
56mussolino,c,etal.(2011)nucleicacidsres.39,9283-9293.
57maeder,m.l.,etal.(2008)mol.cell,31,294-301.
58hwang,w.y.,etal.(2013)plosone,8:e68708.
59feng,z.,etal.(2013)cellres.23,1229-1232.
60mali,p.,etal.(2013)science,339,823-826.
61zhou,j.,etal.(2014)febsj.doi:10.1111/febs.12735.
62fu,y.,etal.(2013)nat.biotechnol.31,822-826.
63fu,y.,etal.(2014)natbiotechnol.doi:10.1038/nbt.2808.
64hsu,p.d.,etal.(2013)nat.biotechnol.31,827-832.
65sander,j.d.,etal.(2007)nucleicacidsres.35,w599-605.
66sander,j.d.,etal(2010)nucleicacidsres.38,w462-468.
- 还没有人留言评论。精彩留言会获得点赞!