一种百合的易感真菌基因LrWRKY-S1及其应用的制作方法
本发明属于植物基因工程技术领域,特别的涉及一种百合的易感真菌基因lrwrky-s1及其应用。
背景技术:
在农业生产中,植物病害尤其是真菌病害一直是限制农作物产量提高的重要因素。真菌病原体和植物宿主间的作用可根据真菌感染如何进展而分为“抗性”或“易感性”,它们涉及不同的宿主细胞基因和调节所述宿主细胞基因表达的机制。在抗性作用中,生物体通过产生抗真菌蛋白(afp),抗真菌肽或化合物来防治真菌侵袭。抗真菌蛋白是先天性免疫反应中的进化保守组件,并且是主要的效应分子。抗真菌蛋白参与生物体的组成抗性或者诱导抗性机制,使生物体能够有效地抵抗致病真菌的侵害。例如编码植物抗毒素、抗微生物素和致病相关(pr)蛋白的基因。相反,易感性作用是感染改变宿主细胞的基因表达,通过抑制植物抗病激素信号通路、抗毒素或致病相关蛋白的表达,或者提供专性植物病原体生物营养生成所必需的基因产物来实现,可以将真菌的易感性基因通过分子手段改良植物抵抗真菌病害能力或制备生物抗真菌诱导剂,以及将真菌的易感性基因导入杂草中使其更容易感病进而逐渐消亡。
但目前,研究热点主要是从许多种动植物、真菌及细菌中分离出来抗真菌蛋白和抗真菌肽,再将抗真菌蛋白基因引入农作物后,来显著增强其对致病真菌的抗性。如发明专利cn201810451848.4,公开了一种假结核耶尔森氏菌抗真菌蛋白,为农作物、蔬菜及水果的生物防治提供一种新的抗真菌蛋白基因,可用于农业、制药或防腐中对抗真菌疾病。发明专利cn201811236660.4公开一种抗真蛋白β-1,3-葡聚糖酶基因,该β-1,3-葡聚糖酶能有效的水解真菌的细胞壁,如安琪酵母和稻瘟病菌,进而抑制稻瘟孢子的萌发和附着孢的形成,展现出良好的抗真菌活性。利用基因生产的酶制剂可用于农业上植物病原真菌的生物防治。发明专利cn200910210538.4公开了一种提高小麦纹枯病抗性的方法,通过采用基因工程方法将天麻的抗真菌蛋白基因和兔防御素基因导入常规小麦并表达,增加小麦的纹枯病抗性。根据抗真菌蛋白的结构和/或功能,抗真菌蛋白可以分为:包括几丁质酶和几丁质酶样蛋白,几丁质结合蛋白,亲环蛋白样蛋白,防御素和防御素样蛋白,脱氧核糖核酸酶,胚胎丰富蛋白,葡聚糖酶,凝集素,脂质转移蛋白,过氧化物酶,蛋白酶抑制剂,核糖核酸酶,核糖体失活蛋白,储存2s白蛋白和类似丘脑蛋白等抗真菌蛋白。而现有技术中关于易感基因的研究和分离较少,因此,开发新的、有效真菌易感基因为生态农业的发展提供了理论依据,拓宽了植物病害防治的思路和在提高农业生产的经济效益方面有重要意义。
岷江百合(liliumregalewilson)是百合科百合属中重要的野生种质资源,广泛分布于我国四川岷江流域(杨利平和符勇耀著,中国百合资源利用研究[m],哈尔滨:东北林业大学出版社,2018.11)。相对于模式植物,岷江百合的生长发育调控机制具有复杂性和特殊性。前人研究表明,岷江百合对百合真菌病和病毒病具有较强的抗性,目前已经从岷江百合中获得多个逆境胁迫相关基因,如lr14-3-3、lrpr10、lrbzip1和lrwrky1等(lietal.,scihort2014,168:9-16;heetal.,genesgenom2014,36:497-507;zhangetal.,genesgenom2014,36:789-798;hanetal.,scihort2016,198:370-378;专利号:201610001896.4)。岷江百合(liliumregalewilson)的基因组测序尚未完成,课题组前期利用灰葡萄孢侵染岷江百合(现蕾期)叶片,提取处理组和对照组总rna,送华大基因进行rna测序。结果表明,大部分lrwrky基因表现出显著上调表达,而有一个lrwrky基因只有轻微的响应,猜测该基因可能有着不同的基因功能。目前,有关岷江百合中的真菌的易感基因研究鲜有报道。
技术实现要素:
针对现有技术存在的空白,本发明的目的在于提供一种百合的易感真菌基因lrwrky-s1及其应用,为植物的遗传改造提供了一个新的易感真菌基因。
本发明的另一个目的在于提供一种百合的易感真菌基因lrwrky-s1在植物培育中的应用,可以通过分子手段改良植物抵抗真菌病害能力或制备生物抗真菌诱导剂,以及将该基因导入杂草中使其更容易感病进而逐渐消亡提供理论基础和技术支持。
为实现上述目的,本发明采用如下技术方案:一种百合的易感真菌基因lrwrky-s1,其核苷酸序列如seqidno.1所示或具有如seqidno.1所示的核苷酸序列经一个或多个核苷酸的替换、缺失或插入得到的具有相同功能的核苷酸序列。
本发明还提供了上述百合的易感真菌基因lrwrky-s1编码的蛋白,其氨基酸序列如seqidno.2所示或具有如seqidno.2所示的氨基酸序列经一个或多个氨基酸的替换、缺失或插入得到的具有相同功能的氨基酸序列。
本发明还提供了一种含有上述百合的易感真菌基因lrwrky-s1的植物表达载体。
本发明还提供了含有上述百合的易感真菌基因lrwrky-s1的宿主细胞。
本发明还提供了上述百合的易感真菌基因lrwrky-s1在培育植物对真菌敏感性或抗性方面的应用。作为优选的,所述真菌为灰葡萄孢、尖孢镰刀菌和炭疽病菌中的至少一种。
本发明的另一个目的在于提供一种增加植物抗病性的方法,所述方法包括将植物中所述基因lrwrky-s1或其同源基因的敲除或抑制表达。
本发明的另一个目的在于提供一种增加植物致病性的方法,其特征在于,所述方法包括向植物细胞中导入含有上述基因lrwrky-s1的植物表达载体或宿主细胞,使基因lrwrky-s1过表达。
进一步,所述植物对灰葡萄孢、尖孢镰刀菌或炭疽病菌产生抗病性或致病性。作为优选的,所述植物为单子叶植物或双子叶植物。进一步,所述单子叶植物为百合属植物;双子叶植物为拟南芥、烟草等。
相比现有技术,本发明具有如下有益效果:
1、本发明首次克隆了岷江百合lrwrky-s1基因全长序列,获得其编码的蛋白序列,并将该蛋白在拟南芥中过表达后,通过抑菌实验,研究该蛋白的生物学功能,实验结果表明该蛋白使得植物更容易感染病原真菌,如灰葡萄孢(botrytiscinerea)、尖孢镰刀菌(fusariumoxysporum)和炭疽病菌(colletotrichumliliacearum)等。该研究为植物基因改造工程提供了一个高效的感病基因,并为进一步培育具有真菌抗性或敏感性植物的新材料或新品种奠定了基础,同时也拓宽了植物病害防治的思路,具有良好的应用前景。
2、本发明通过功能基因组学研究证明lrwrky-s1基因与其它lrwrky家族基因(如:lrwrky1-4,专利申请号为201610001896.4,201911106106.9,201911106095.4和201911105589.0)功能不同,它是一个感病基因,提供一种改良植物真菌病抗性的新方法和新途径。通过分子检测技术发现,lrwrky-s1基因在兰州百合、龙牙百合和卷丹百合中的同源基因表达水平较高,因此,通过分子手段如定向敲除或反向抑制等可以定向培育百合抗病新品种,或者采用病毒诱导的基因沉默(vigs)和rna干扰的瞬时抑制表达方法制备抗真菌诱导剂来提高百合抗病性。上述方法具有育种周期短,操作简单,容易获得目标材料等优点。利用该方法获得如观赏花卉和农作物等新材料,可以减少农药使用,降低环境污染。另外,也可以利用该基因制备易感的植物材料,降低其抗病性,如转入杂草中等。本发明为今后利用该基因进行植物分子遗传改良提供理论和技术支持,因此,具有重要的推广价值。
附图说明
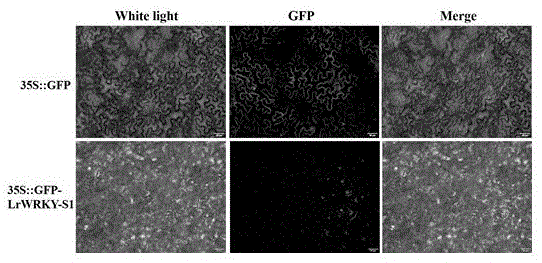
图1为岷江百合lrwrky-s1转录因子的亚细胞定位分析;上组图从左到右依次分别为对照组(35s::gfp)在白光下明场图、激发光下暗场图和组合图;下组图从左到右依次分别为实验组(35s::gfp-lrwrky-s1)在白光下明场图、激发光下暗场图和组合图。
图2为lrwrky-s1转基因拟南芥基因组dna的pcr检测结果电泳图,其中marker:dl2000dnamarker(大连宝生物);泳道1-20,23,24为lrwrky-s1转基因株系,泳道21为野生型(wt),22为阴性对照。
图3为转基因拟南芥中lrwrky-s1基因的定量表达分析图;其中,wt为野生型拟南芥对照,ox-l5、ox-l7、ox-l23和ox-l24分别为转基因拟南芥阳性株系。
图4为转基因拟南芥离体叶片抑菌活性效果图;图中a、c、e为野生型拟南芥叶片;b、d、f为lrwrky-s1转基因拟南芥代表的叶片;其中a和b接种灰葡萄孢菌,c和d接种尖孢镰刀菌,e和f接种炭疽病菌。
图5为wrky-s1基因在不同百合中的转录表达水平表达分析;a代表不同百合幼苗叶片组织,b代表不同百合鳞茎组织,其中,1为岷江百合,2为卷丹百合,3为兰州百合,4为龙牙百合。
具体实施方式
下面结合具体实施例和附图对本发明作进一步详细说明,但本发明保护范围不局限于所述内容。实施例中所述原料如无特殊说明,均为普通市售产品。实施例中所述的实验方法无特别说明,即按常规分子生物学实验方法操作。
实施例1岷江百合lrwrky-s1基因克隆与序列分析
以岷江百合叶片为材料,采用trizoltmplusrnapurificationkit(12183555,invitrogentm),按说明书步骤提取总rna,利用dnasei(18047019,invitrogentm)去除残余的微量dna,并用分光光度计测定rna的浓度,备用。
取约2.0μg岷江百合叶片总rna,按照primescriptiifirst-strandcdnasynthesiskit(6210a,takara)说明书步骤合成第一链cdna。
pcr扩增体系:2xfastpfumastermax10ul,正向引物(lrwrky-s1-1-f,10μm)1μl,反向引物(lrwrky-s1-r,10μm)1μl,模板(cdna)1μl,无菌ddh2o补足至20μl。
正向引物和反向引物为:
lrwrky-s1-f:5’-gtccgaatatcatggacggag-3’
lrwrky-s1-r:5’-ctacaacatttaaacgaagaaggcag-3’
pcr反应程序为:预变性94℃,3min;94℃,30s;58℃,40s;72℃,1min,38个循环;72℃,10min。
获得pcr产物通过琼脂糖凝胶电泳分析,在1kb左右可以观察到一条特异性的扩增条带。按照胶回收试剂盒(9672,takara)纯化后备用。
利用平末端加a试剂将纯化的dna片段加a,通过ta克隆将其与pmd20-tvector(6019,takara)相连,连接产物转化大肠杆菌dh5a,从含有氨苄霉素(100mg/l)的lb培养板上挑取2-3个阳性克隆测序进行测序分析,结果表明,岷江百合lrwrky-s1基因全长序列如seqidno.1所示,包括984bp的开放阅读框(不含终止密码)和下划线部分的部分5’-utr或3’-utr序列。
根据seqidno.1所示序列,通过dnaman软件翻译,获得岷江百合lrwrky-s1的蛋白质序列如seqidno.2所示,含有328个氨基酸(*表示终止信号)。借助蛋白亚细胞定位在线分析(wolfpsort)显示,lrwrky-s1在植物细胞中的表达定位可能性依次为nucl:12,extr:1,cysk:1。通过cnlsmapper在线预测表明,在lrwrky-s1蛋白的n端具有核定位信号序列(rremskkrktlpkw)。将lrwrky-s1全长核苷酸序列和氨基酸序列在ncbi数据库中进行blast比对,发现其与经过转录组测序分析获得的拼接序列麝香百合llwrky41(genbank:mh614350.1)和岷江百合lrwrky46(genbank:mg149582.1)相似度较高,核苷酸序列相似度为94.22%(930/987)和93.82%(926/987),氨基酸序列相似度为92.07%(302/328)和90.85%(298/328),分别存在26个氨基酸和31个氨基酸序列的区别,从比对结果来看lrwrky-s1基因为新基因。
实施例2gfp融合表达载体构建及亚细胞定位分析
(1)构建ptf101-p35s::gfp-lrwrky-s1表达载体
根据ptf101-gfp载体序列和lrwrky-s1基因全长序列(seqidno.1),设计正向引物(lrwrky-s1-inf-f1)和反向引物(lrwrky-s1-inf-r1)。以实施例1中ta连接的阳性克隆质粒为模板,进行lrwrky-s1基因片段的pcr扩增。
所述引物序列如下:
lrwrky-s1-inf-f1:
5’-
lrwrky-s1-inf-r1:
5’
下划线处为in-fusion克隆载体接头序列,加粗序列为酶切位点。
pcr反应体系:高保真扩增酶primestarhs(r010a,takara)0.5μl,5xprimestarbuffer(mg2+plus)10μl,正向引物(10μm)1μl,反向引物(10μm)1μl,模板(50倍稀释的质粒)1μl,dntp(2.5mm)4μl,无菌ddh2o补足至50μl。
pcr反应条件:预变性95℃,5min;95℃,30s;58℃,40s;72℃,1min,38个循环;72℃,7min。
将pcr扩增产物进行琼脂糖凝胶电泳检测,按照胶回收试剂盒(9672,takara)步骤回收纯化,即获得目的基因片段。
用hindiii和bamhi双酶切处理ptf101-gfp表达载体。酶切体系为:ptf101-gfpvector5μl;hindiii0.5μl;bamhi0.5μl;buffer10xk2μl;无菌ddh2o补足至20μl;37℃反应3h。酶切结束后,根据takara琼脂糖凝胶回收试剂盒回收ptf101-gfp载体片段。
利用无缝克隆技术(in-fusionhdcloningkit,takara)构建ptf101-p35s::gfp-lrwrky-s1表达载体。
重组反应体系为:
purifedpcrfragment(回收的lrwrky-s1目的片段)50ng;linearizedvector(ptf101-gfp载体)100ng;5xin-fusionhdenzymepremix2μl;无菌ddh2o补足至10μl。50℃反应15min。按照分子克隆实验指南将上述重组反应体系转化大肠杆菌dh5a,并涂布于含有壮观霉素(spec,100mg/l)的筛选培养板上,通过阳性克隆测序,获得正确的含lrwrky-s1基因片段的重组表达载体ptf101-p35s::gfp-lrwrky-s1。重组表达载体中报告基因gfp与目的基因lrwrky-s1的5’端融合后,位于组成型启动子p35s下游,形成融合表达;lrwrky-s1的3’端组装有nos终止子,可以有效终止融合基因的转录。报告基因gfp经过蓝光激发,无需辅助因子和底物就能发出绿色荧光,作为报告基因可以检测目的基因表达情况。
(2)lrwrky-s1的亚细胞定位分析
通过常规冻融法将构建的重组表达载体ptf101-p35s::gfp-lrwrky-s1转入农杆菌菌株eha105中,通过pcr筛选阳性克隆。以含有ptf101-gfp质粒的农杆菌菌株为阳性对照,按照霍琳等(农杆菌介导的烟草瞬时表达试验条件优化,分子植物育种,2016,14(1):80-85)方法制备农杆菌注射缓冲液。选择光照培养箱中正常生长具有8-10片叶子完全展开的烟草进行注射,用去除针头的注射器将注射缓冲液缓慢推入叶片背面。然后,把转化后的植株重新放回培养箱中,培养36h-48h后进行观察。
用剪刀小心剪下转化后的烟草叶片放在载玻片上,加1滴蒸馏水,制成装片;然后放在荧光显微镜上,在激发光波长488-507nm的蓝光下,进行荧光观察。结果如图1所示,对照组(图1上)在烟草表皮细胞的细胞核和细胞膜上均有荧光表达,而实验组(图1下)只在烟草表皮细胞的细胞核上特异性表达。以上结果表明,岷江百合lrwrky-s1是1个核定位的转录因子蛋白,与植物中大部分wrky因子表达定位情况相符。
实施例3lrwrky-s1基因过表达载体构建及拟南芥的遗传转化
(1)构建pbi121-p35s::lrwrky-s1过表达载体
根据pbi121载体序列和lrwrky-s1基因序列(seqidno.1),设计引物lrwrky-s1-inf-f2和lrwrky-s1-inf-r2,引物中引入无缝克隆(in-fusion)载体接头序列和酶切位点序列。以实施例1中ta连接的阳性克隆质粒为模板,进行lrwrky-s1基因片段的特异性扩增。
所述引物序列如下:
lrwrky-s1-inf-f2:
5’-ggactctagaggatccgtccgaatatcatggacggag-3’
lrwrky-s1-inf-r2:
5’-gatcggggaaattcgagctcctacaacatttaaacgaagaaggcag-3’
其中,下划线处为in-fusion克隆载体接头序列。
pcr反应体系为:高保真扩增酶primestarhs(r010a,takara)0.5μl,5xprimestarbuffer(mg2+plus)10μl,正向引物(10μm)1μl,反向引物(10μm)1μl,模板(50倍稀释的质粒)1μl,dntp(2.5mm)4μl,无菌ddh2o补足至50μl。
pcr反应条件:预变性95℃,5min;95℃,30s;58℃,40s;72℃,1min,38个循环;72℃,10min。
将pcr扩增产物进行琼脂糖凝胶电泳检测。扩增得到的目的片段与预期片段大小相同,按照胶回收试剂盒(9672,takara)的说明书步骤回收纯化,即获得目的基因片段。
用bamhi和saci双酶切处理pbi121植物双元表达载体。酶切体系为:pbi121vector5μl;bamhi0.5μl;saci0.5μl;buffer10xk1μl;无菌ddh2o补足至20μl;37℃反应3h。酶切结束后,根据takara琼脂糖凝胶回收试剂盒回收pbi121载体大片段。
利用无缝克隆技术(in-fusionhdcloningkit,takara)构建pbi121-p35s::lrwrky-s1重组表达载体。
重组反应体系为:
purifedpcrfragment(回收的lrwrky-s1目的片段)50ng;linearizedvector(pbi121)100ng;5xin-fusionhdenzymepremix2μl;无菌ddh2o补足至10μl。50℃反应15min。然后按照分子克隆实验指南将上述重组反应体系转化大肠杆菌dh5a,并涂布于含有卡纳霉素(100mg/l)的筛选培养板上,通过阳性克隆筛选和测序,获得正确的含lrwrky-s1基因片段的重组表达载体pbi121-p35s::lrwrky-s1。重组表达载体中目的基因lrwrky-s1的5’端位于组成型启动子p35s下游,它能使lrwrky-s1基因过量表达;lrwrky-s1的3’端组装有nos终止子,可以有效终止基因的转录。在重组表达载体上组装有nptii基因,作为转基因植物的筛选标记,可以用卡纳霉素进行转基因植株的筛选。在重组表达载体上组装有lb和rb序列,促使组装于其间的表达框架和筛选标记基因nptii整合至植物受体染色体上。
(2)拟南芥的遗传转化
拟南芥的遗传转化采用浸花法(zhangx.,etal.,natprotoc.2006,1:641-646)。将携带pbi121-p35s::lrwrky-s1载体的农杆菌导入哥伦比亚col型拟南芥。用卡那霉素(100mg/l)筛选具抗性的拟南芥再生幼苗,通过常规pcr方法扩增lrwrky-s1目标基因。
pcr扩增引物为:
lrwrky-s1-f:5’-gtccgaatatcatggacggag-3’
lrwrky-s1-r:5’-ctacaacatttaaacgaagaaggcag-3’
pcr检测结果如图2所示,在野生型植株(wt)中检测不到lrwrky-s1基因的存在,只有在转基因株系(ox-l5、ox-l7、ox-l23和ox-l24)中扩增出明显的条带,而在其他的转基因株系中扩增的条带不明显,表明至少有4个阳性株系中,重组表达框已导入拟南芥基因组。
进一步,采用trizol试剂(invitrogentm),按说明书步骤提取野生型和转基因阳性拟南芥叶片总rna,利用dnasei(invitrogentm)去除残余dna,采用cdna反转录试剂(6210a,takara)并按照说明书步骤合成第一链cdna。以拟南芥actin基因(at3g53750)为内参,通过定量rt-pcr方法分析转基因阳性株系与野生型中lrwrky-s1基因的表达水平。
检测actin基因引物为:
actin-qf:5'-gtctggattggaggatccat-3'
actin-qr:5'-ccggtgaacaatcgacgggc-3'
检测lrwrky-s1基因引物为:
lrwrky-s1-qf:5'-ccgcaacaccatcagccaga-3'
lrwrky-s1-qr:5'-ctgaggccgctgacgagatg-3'
相对表达水平采用2-δδct方法(livakkj,schmittgentd,2001.analysisofrelativegeneexpressiondatausingrealtimequantitativepcrandthe2(-deltadeltac(t))method.methods25,402–408)分析计算。结果如图3所示,与野生型植株(wt)相比,在4个代表转基因株系(ox-l5、ox-l7、ox-l23和ox-l24)中目的基因lrwrky-s1均显著上调表达,而在野生型植株(wt)中检测不到lrwrky-s1的表达,表明lrwrky-s1已经导入拟南芥基因组并成功转录表达。
实施例4:lrwrky-s1转基因阳性株系的真菌抗性分析
将病原真菌灰葡萄孢、尖孢链刀菌和炭疽病菌分别接种到pda培养基(200g/l马铃薯,15g/l琼脂,20g/l葡萄糖)中,在28℃黑暗条件下培养1周。以温室中正常生长4周左右的野生型和转基因拟南芥为材料,利用打孔器将大小相等的新鲜真菌菌丝块分别接种到离体的莲座叶片上,在培养皿中保持湿度,放于光照培养箱正常培养,2d后观察病原真菌的侵染情况。结果如图4所示。
从图中可以看出,野生型拟南芥(wt)的叶片在接种3种不同病原真菌后,受伤害的病斑面积不明显,其叶脉组织中的颜色有改变(图4a、图4c和图4e);而转lrwrky-s1基因株系的叶片(图4b、图4d和图4f)接种真菌以后,其受伤害的病斑面积更加严重,颜色改变也更加明显。因此表明,过表达lrwrky-s1基因会使转基因材料对灰葡萄孢、尖孢镰刀菌和炭疽病菌等真菌的抗性显著降低,表现出更加敏感的表型。
实施例5:wrky-s1基因在不同百合中的表达水平分析
以4种百合(岷江百合、卷丹百合、兰州百合和龙牙百合)的幼苗叶片和鳞茎为材料,采用minibestplantrnaextractionkit(9769,takara)从叶片组织提取总rna,采用rnaisoforpolysaccharide-richplanttissue(9752a,takara)从鳞茎组织提取总rna,利用primescripttmrtreagentkitwithgdnaeraser(pr047a,takara)反转录获得cdna,以百合18s基因为内参,采用半定量rt-pcr方法分析wrky-s1基因在不同百合中的表达水平。
检测18s基因引物为:
18s-f:5’-cgcaaggctgaaacttaaagg-3’
18s-r:5’-cagacaaatcgctccaccaac-3’
检测lrwrky-s1基因的引物为:
wrky-s1-sf:5’-atggacggaggctctgg-3’
wrky-s1-sr:5’-cgaagaaggcagaggcatcg-3’
pcr反应体系为:taqdnapolymerase(r001b,takara)0.5μl,10xpcrbuffer(mg2+plus)5μl,正向引物(10μm)1μl,反向引物(10μm)1μl,cdna模板1μl,dntp(2.5mm)4μl,无菌ddh2o补足至50μl。
pcr反应条件:预变性95℃,5min;95℃,30s;58℃,40s;72℃,1min,28个循环;72℃,10min。
将pcr扩增产物进行琼脂糖凝胶电泳检测。结果如图5所示,4种不同百合的叶片组织(图5a)中,兰州百合和龙牙百合的wrky-s1表达水平最高(图5a-3,图5a-4),其次是卷丹百合(图5a-2),表达水平最低的是岷江百合(图5a-1);在鳞茎组织中(图5b),兰州百合的wrky-s1表达水平最高(图5b-3),其次是卷丹百合和龙牙百合(图5b-2,5b-4),表达水平最低的也是岷江百合(图5b-1)。以上结果与岷江百合对真菌的高抗性相符,与其它3种百合对真菌抗性较差相一致。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
sequencelisting
<110>长江师范学院;
<120>一种百合的易感真菌基因lrwrky-s1及其应用
<160>16
<170>siposequencelisting1.0
<210>1
<211>1007
<212>dna
<213>岷江百合(liliumregalewilson)
<400>1
gtccgaatatcatggacggaggctctgggacagtgcagactctgctgatccacgagctca60
cccgaggccatgaactcctcggtcagctcttggccaatctcgatcaaccaacggtaacca120
acccctgcaaacaactggcatcagagatactcttatctatcgagagggcactctgcatag180
ccaaagcaagcatcctcgaaagcccccgcctgaacccctcctatggaggcaccgactcca240
gtgtcagccctcgcagtgagagctccgagcaggcctccaaggagctcgagcgcagagaga300
tgtcgaaaaagaggaaaacactacccaaatggaccaaccaggtgctggcaggagcaggag360
aaaggccggagggaccggcggacgatgggtacagctggagaaagtacggtcagaaagaaa420
ttctcggtgcgaaacatccaaggggctactaccgttgcacgcacagaaatacccgcggct480
gcttggcaaccaaacaagtacagcggtccgatcagaacccgtcgatattggacatcacct540
atcgaggtgatcacacttgccaccagaagcagcagctagtttttccccgcggggagaccg600
agcaggagcaggagcaggagcagggccaggagattcatcccctgatggaccgtgttcagc660
cgcaacaccatcagccagaattgctgctgagcttccagaccggcctcagagtcaggacca720
ctggcagctgcattaagaccgataatatggtcttctcctcgcctttggggatggagaatt780
gcttcaatagcaatttttcgccttccttcatctcgtcagcggcctcagagtccaactttg840
ggggtgctaatccacagggtgcagagtctgactataaagaaatcgtcactgcagcaagct900
cagcggtcgattcacctttcgtagacatggatttcatgctcggcaacatagatttcgacc960
cagacttccacttcgatgcctctgccttcttcgtttaaatgttgtag1007
<210>2
<211>328
<212>prt
<213>岷江百合(liliumregalewilson)
<400>2
mdggsgtvqtlliheltrghellgqllanldqptvtnpckqlaseillsieralciakas60
ilesprlnpsyggtdssvsprsesseqaskelerremskkrktlpkwtnqvlagagerpe120
gpaddgyswrkygqkeilgakhprgyyrcthrntrgclatkqvqrsdqnpsildityrgd180
htchqkqqlvfprgeteqeqeqeqgqeihplmdrvqpqhhqpelllsfqtglrvrttgsc240
iktdnmvfssplgmencfnsnfspsfissaasesnfgganpqgaesdykeivtaassavd300
spfvdmdfmlgnidfdpdfhfdasaffv328
<210>3
<211>21
<212>dna
<213>人工序列
<400>3
gtccgaatatcatggacggag21
<210>4
<211>26
<212>dna
<213>人工序列
<400>4
ctacaacatttaaacgaagaaggcag26
<210>5
<211>43
<212>dna
<213>人工序列
<400>5
ttccccgggctcgagaagcttatggacggaggctctgggacag43
<210>6
<211>46
<212>dna
<213>人工序列
<400>6
ttatctagatccggtggatccttaaacgaagaaggcagaggcatcg46
<210>7
<211>37
<212>dna
<213>人工序列
<400>7
ggactctagaggatccgtccgaatatcatggacggag37
<210>8
<211>46
<212>dna
<213>人工序列
<400>8
gatcggggaaattcgagctcctacaacatttaaacgaagaaggcag46
<210>9
<211>20
<212>dna
<213>人工序列
<400>9
gtctggattggaggatccat20
<210>10
<211>20
<212>dna
<213>人工序列
<400>10
ccggtgaacaatcgacgggc20
<210>11
<211>20
<212>dna
<213>人工序列
<400>11
ccgcaacaccatcagccaga20
<210>12
<211>20
<212>dna
<213>人工序列
<400>12
ctgaggccgctgacgagatg20
<210>13
<211>21
<212>dna
<213>人工序列
<400>13
cgcaaggctgaaacttaaagg21
<210>14
<211>21
<212>dna
<213>人工序列
<400>14
cagacaaatcgctccaccaac21
<210>15
<211>17
<212>dna
<213>人工序列
<400>15
atggacggaggctctgg17
<210>16
<211>20
<212>dna
<213>人工序列
<400>16
cgaagaaggcagaggcatcg20
- 还没有人留言评论。精彩留言会获得点赞!