一种高延展性的高性能半导体共轭聚合物及其制备方法
本发明属于有机半导体共轭聚合物领域,具体涉及一种高延展性的高性能半导体共轭聚合物及其制备方法,通过精确调控侧链中直链烷基和硅氧烷链的长度来达到高延展性的高性能半导体共轭聚合物材料的目的。
背景技术:
可穿戴和植入设备的未来前景使可拉伸有机半导体的研究逐渐深入。目前,共轭聚合物中研究最多的是给体-受体(d-a)共轭聚合物,其半导体材料本身具有一定的延展性,传统的烷基侧链的半导体共轭聚合物薄膜延展后电学性能不佳,且文献中报道的本征可拉伸共轭聚合物材料种类有限。因此,急需开发高延展性的高性能半导体共轭聚合物。开发此类的材料所面临的挑战源于高电学性能所需要的结晶性和高拉伸性所需要的无序性的矛盾的材料设计规则。共轭聚合物中的分子间π-π堆积通常会导致高结晶域,这对于高载流子传输是有利的,然而,这种半节晶形态使共轭聚合物膜变脆,这不利于机械变形。
有机硅聚合物在工程应用中非常广泛,其具有高度柔性,例如,聚二甲基硅氧烷(pdms)具有任何已知聚合物中最低的玻璃化转变温度(≈150k),由于硅氧键长和键角大于碳碳键的键长和键角,可以增加侧链柔软性并形成弹性缓冲区构筑可变形的非晶区域,降低聚合物薄膜的相对结晶度。传统较多使用的七甲基三硅氧烷的侧链较短,无法形成可应变的弹性区域。因此,若将精确调节的烷基直链和长硅氧烷基团引入共轭聚合物体系中,形成并利用可变形的非晶区域实现拉伸过程中的应力耗散,通过有机硅氧烷侧链包覆的半结晶聚集体结连接链内和链间电荷传输,得到兼具高延展性和电荷传输特性的半导体共轭聚合物。
技术实现要素:
本发明是利用延长的有机硅基团提供的超柔软性,提供一种高延展性的高性能半导体共轭聚合物及其制备方法。本发明聚合物具有精确长度的烷基直链和两种可变换的有机硅氧烷侧链。本发明结构设计多样化的目的在于精确调节聚合物延展性和电学性能,达到最优的组合。本发明的共轭聚合物半导体材料有望实现在柔性或拉伸电子器件中的应用。
本发明是通过以下技术方案来实现的:
第一方面,利用烷基直链的长度调节主链和侧链之间的距离,保证最佳的电学性能,较短的烷基侧链会导致聚合物较低的溶解度,导致半导体材料无法溶液合成和加工;较长的烷基侧链会导致聚合物材料较低的电荷传输特性。m为5~9之间聚合物材料拥有较优异的电学性能。
另一方面,利用可调节的有机硅氧烷基团作为柔性基团,短的有机硅氧烷基团很难溶液加工且其薄膜延展性很差,较长的有机硅氧烷基团拥有足够的延展性,但由此制备的聚合物材料电荷传输性能很差,无法在器件中正常使用。p1为4~7,p2为2~5之间相对应的聚合物材料拥有较优异的延展性同时保持较高的电学性能。
本发明高延展性的高性能半导体共轭聚合物,是基于精确调节的烷基直链以及可调节的有机硅氧烷基团作为侧链r的半导体共轭聚合物,其结构通式如下所示:
上述结构通式中,主链的共轭骨架可以多样化调整。其中a为缺电子受体单元,可由吡咯并吡咯二酮(dpp)单元、双(7-氮杂-3-亚乙基)-苯并二呋喃二酮、异靛蓝、ndi中的任一种形成;d为富电子给体单元,可由单噻吩、单硒吩、联噻吩、并二噻吩、并三噻吩中任一种形成。
上述结构通式中,r为侧链,选自如下结构中的一种:
其中m=5~9,p1=4~7,p2=2~5。
本发明选用最佳的烷基直链和有机硅氧烷链组合共同构筑可变形的非晶区域,实现拉伸过程中的应力耗散,同时调整共轭骨架结构,得到兼具高延展性和电荷传输特性的聚合物。本发明通过精确调控侧链中直链烷基和硅氧烷链的长度来精确调控聚合物侧链的体积大小,最终精确调节聚合物薄膜的相态分布,利用有机硅氧烷侧链作为弹性缓冲区构筑可变形的非晶区域,实现拉伸过程中的应力耗散,进而达到半导体材料高延展性的目的。
本发明高延展性的高性能半导体共轭聚合物可采用两种方式制备:第一,采用直链烷基链加直链型有机硅氧烷链作为侧链构成最终分子结构;第二,采用直链烷基链加分叉型有机硅氧烷链作为侧链构成最终分子结构。
本发明高延展性的高性能半导体共轭聚合物的制备方法,包括如下步骤:
步骤1:受体单元的烷基化
以卤化烷基直链烯与受体单元为原料,在k2co3的存在下于溶剂中进行卤代反应,反应温度110-120℃,反应获得连接了烷基直链的受体单元;
步骤2:硅氧烷的加成
以步骤1获得的连接了烷基直链的受体单元和有机硅氧烷为原料,在催化剂的存在下于溶剂中进行加成反应,反应温度为50-55℃,反应获得连接了侧链的受体单元;
步骤3:溴化反应
将步骤2获得的连接了侧链的受体单元与nbs(n-溴代琥珀酰亚胺)在溶剂中进行溴化反应,室温下反应获得溴化的受体单元;溴化反应中可以加入自由基捕获剂(tempo),能够获得高产率的溴化产物;
步骤4:偶联
将步骤3获得的溴化的受体单元与给体单元进行偶联反应,偶联反应在催化剂和配体的存在下于溶剂中进行,于120-140℃下反应获得目标聚合物。
步骤1中,所述卤化烷基直链烯为c7-c11的直链烯烃。
步骤1中,所述受体单元包括吡咯并吡咯二酮(dpp)、双(7-氮杂-3-亚乙基)-苯并二呋喃二酮、异靛蓝、ndi中的任一种。
步骤1中,卤化烷基直链烯与受体单元的反应当量比为1:2-2.5。
步骤1中,所述溶剂优选为dmf。
步骤2中,所述有机硅氧烷包括精确控制长度的直链或分叉有机硅氧烷,其结构通式如下所示:
其中p1=4~7,p2=2~5。
所述有机硅氧烷可以市购获得,也可以自制。自制方法参考acsappl.mater.interfaces2020,12,41832-41841。
步骤2中,所述催化剂为karstedt催化剂(二甲苯中的铂二乙烯基四甲基硅氧烷络合物,3wt%)。
步骤2中,连接了烷基直链的受体单元和有机硅氧烷的反应当量比为1:2-2.5。
步骤2中,所述溶剂优选为甲苯。
步骤3中,连接了侧链的受体单元与nbs的反应当量比为1:2-2.5。
步骤3中,所述溶剂优选为氯仿。步骤3中,获得的溴化产物为双溴化的受体单元。
步骤4中,所述给体单元包括单噻吩、单硒吩、联噻吩、并二噻吩、并三噻吩中任一种。
步骤4中,所述催化剂为三(二亚苄基丙酮)二钯;所述配体为三(邻甲基苯基)磷;所述溶剂包括氯苯、甲苯等。
步骤4中,溴化的受体单元与给体单元的反应当量比为1:1;催化剂和配体的投料比为受体或给体当量的4%和16%。
本发明的反应路线如下所示:
本发明首先引入精确长度的卤化烷基直链烯到共轭聚合物的受体上作为侧链的一部分,通过调控侧链中烷基直链的长度,可以控制侧链与主链之间的距离,促使主链紧密排列,使分子堆积更加紧密,从而提高聚合物的电荷传输性能;其次利用硅氢加成反应将直链烷基和精确控制长度的直链或分叉有机硅氧烷链连接到共轭聚合物受体上,通过调控侧链中的直链或分叉有机硅氧烷链的长度以及硅原子数精确控制侧链的弹性体积,使构筑的弹性非晶区域控制在合理的区间,从而达到制备高延展性的高性能半导体材料的目的;最后用双锡化的富电子给体单元(d)和连接了硅氧烷柔性基团的双溴化缺电子受体单元(a)在stille反应条件下共聚,用甲醇沉淀,然后索氏提取提纯共轭聚合物。
本发明的有益效果体现在:
1、本发明新型半导体共轭聚合物是一种低模量、高延展性的高性能半导体共轭聚合物,聚合物主链可以多样化调节,以可调节的烷基直链和有机硅氧烷基团为侧链。重点在于共轭聚合物中引入可调节的有机硅氧烷基团的侧链可以构筑可变形的非晶区域,降低聚合物的弹性模量,利用变形的非晶区域实现拉伸过程中的应力耗散,提高拉伸性,同时调节主链结构,通过有机硅氧烷侧链包覆的半结晶聚集体结连接链内和链间电荷传输,获得高性能的半导体聚合物。
2、本发明半导体共轭聚合物可以作为空穴型电荷传输材料应用于柔性电子器件中。
3、本发明半导体共轭聚合物具有极好的拉伸下的电学性能,将其薄膜转移至pdms上进行拉伸后制作的场效应晶体管器件具有很高的传输性能。本发明对于设计高延展性的高性能本征半导体共轭聚合物材料具有很高的指导意义。
附图说明
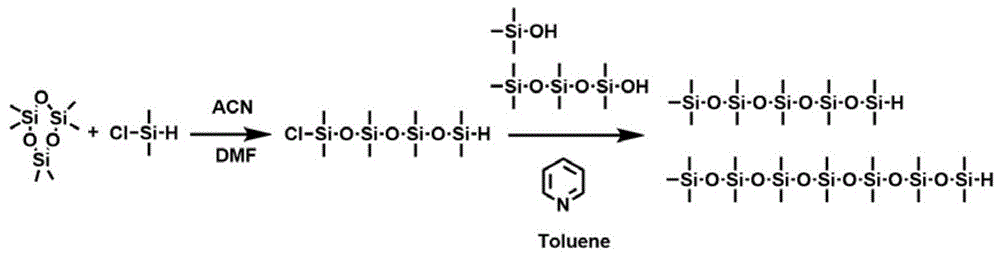
图1为部可调节长度的直链有机硅氧烷基团的侧链的合成路径示意图。
图2为高产率的nbs溴化的合成路径图。
图3为基于dpp双溴单体和噻吩双锡化单体的半导体共轭聚合物的合成路径示意图。
图4为实施例1中半导体共轭聚合物ptdpptt-c6-si5(m=6,p1=4)薄膜在pdms上进行不同应变拉伸后转移到底栅顶接触的器件上的转移曲线。
图5为实施例2中半导体共轭聚合物ptdpptt-c6-si7(m=6,p1=6)薄膜在pdms上进行不同应变拉伸后转移到底栅顶接触的器件上的转移曲线。
图6为实施例1和实施例2半导体共轭聚合物ptdpptt-c6-si5(m=6,p1=4),ptdpptt-c6-si7(m=6,p1=6)薄膜在pdms上进行拉伸后转移到硅片上测试的afm。
图7为实施例2中半导体共轭聚合物ptdpptt-c6-si7(m=6,p1=6)薄膜在水面自由拉伸下的应力应变曲线和拟合后得出的薄膜模量。
图8为实施例3中半导体共轭聚合物ptdppse-c7-si5(m=7,p2=2)薄膜在pdms上进行不同应变拉伸后转移到底栅顶接触的器件上的转移曲线。
具体实施方式
下面结合附图对本发明的实施例作详细说明。本实施例在以本发明技术方案为前提下进行实施,结合了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
本发明高延展性的高性能半导体共轭聚合物的结构通式如下所示:
其中:m为5~9,p1为4~7,p2为2~5。
实施例1:合成聚合物ptdpptt-c6-si5(m=6,p1=4,主链结构为dpp-并噻吩)
1、如图1所示,本发明所述的方法中部分半导体共轭聚合物的有机硅氧烷基团的合成方法为:以六甲基环三硅氧烷(0.25mol)和氯代二甲基硅氢(0.25mol)为原料,以乙腈(40ml)为溶剂,dmf(2ml)为催化剂,在室温下反应72h,得到氯代八甲基硅氢;在冰浴,氮气通入条件下,将氯代八甲基硅氢(0.045mol)加入到干燥甲苯(40ml)中,冰浴3-5min,加入吡啶(5ml),1-2min内滴加完毕,将三甲基硅醇溶于甲苯,15min内逐渐滴加进反应瓶内,室温反应2-3h,减压蒸馏得到有机硅氧烷(1,1,1,3,3,5,5,7,7,9,9-十一甲基五硅氧烷)。
2、加入k2co3、dpp、6-溴-1-己烯,到单口反应瓶中,加入适量dmf溶解,116℃反应过夜,旋干dmf,进行硅胶色谱,得到深红色的单体dpp-c6。
3、在氮气保护下将步骤2接完双键的化合物dpp-c6溶解在无水甲苯(20ml)中,通过无菌注射器注入十五甲基硅氢,然后加入一滴karstedt催化剂(二甲苯中的铂二乙烯基四甲基硅氧烷络合物,3wt%),将混合物在53℃和氮气氛围中搅拌过夜;将产物进行硅胶色谱,用乙酸乙酯/石油醚(1:50-1:200)作为洗脱液,得到深红色产物dpp-c6-si5。
4、如图2所示,在100ml反应瓶中加入0.38mmol步骤3获得的dpp-c6-si5,加入15ml的氯仿溶解,加入0.1g自由基捕获剂(tempo),然后将nbs(0.8mmol)溶于氯仿中,逐滴加入到反应瓶中,20min内滴加完成,将反应瓶移至室温,过夜反应;将反应后的产物加水萃取2次,加入无水硫酸镁干燥,过滤后得到紫红色的油状物溴化单体;将产物进行硅胶色谱,用石油醚/乙酸乙酯(100:1)作为洗脱液,得到紫红色的双溴单体dpp-c6-si5。
5、如图3所示,本实施例中聚合物ptdpptt-c6-si5(图3中m=6,p1=4)的合成方法如下:
在100ml反应瓶中加入(0.089mmol)双溴单体dpp-c6-si5和0.089mmol并噻吩双锡单体,再加入10ml氯苯,用氮气置换40分钟,再加入0.0045mmol催化剂三(二亚苄基丙酮)二钯,以三(邻甲基苯基)磷0.0045mmol为配体,于130℃反应72小时,将反应冷却至室温,加入70ml甲醇沉淀,过滤固体,固体用丙酮索氏提取12小时,再用正己烷,二氯甲烷提取,旋干得到深紫色的聚合物ptdpptt-c6-si5。
实施例2:合成聚合物ptdpptt-c6-si7(m=6,p1=6,主链结构为dpp-并噻吩)
1、如图1所示,以六甲基环三硅氧烷(0.25mol)和氯代二甲基硅氢(0.25mol)为原料,以乙腈(40ml)为溶剂,dmf(2ml)为催化剂,在室温下反应72h,得到氯代八甲基硅氢;在冰浴,氮气通入条件下,加入氯代八甲基硅氢(0.045mol)加入到干燥甲苯(40ml)中,冰浴3-5min,加入吡啶(5ml),1-2min内滴加完毕,将六甲基硅醇溶于甲苯,15min内逐渐滴加进反应瓶内,室温反应2-3h,减压蒸馏得到最终的有机硅氧烷基团(1,1,1,3,3,5,5,7,7,9,9-十五甲基五硅氧烷)。
2、加入k2co3、dpp、6-溴-1-己烯到单口反应瓶中,加入适量dmf溶解,116℃反应过夜,旋干dmf,进行硅胶色谱,得到深红色的单体dpp-c6。
3、在氮气保护下将步骤2接完双键的化合物dpp-c6溶解在无水甲苯(20ml)中,通过无菌注射器注入十五甲基硅氢,然后加入一滴karstedt催化剂(二甲苯中的铂二乙烯基四甲基硅氧烷漯河物,3wt%),将混合物在53℃和氮气氛围中搅拌过夜;将产物进行硅胶色谱,用乙酸乙酯/石油醚(1:50-1:200)作为洗脱液,得到深红色产物dpp-c6-si7。
4、如图2所示,在100ml反应瓶中加入0.38mmol步骤3获得的dpp-c6-si7,加入15ml的氯仿溶解,加入0.1g自由基捕获剂(tempo),然后将nbs(0.8mmol)溶于氯仿中,逐滴加入到反应瓶中,20min内滴加完成,将反应瓶移至室温,过夜反应;将反应后的产物加水萃取2次,加入无水硫酸镁干燥,过滤后得到紫红色的油状物溴化单体;将产物进行硅胶色谱,用石油醚/乙酸乙酯(100:1)作为洗脱液,得到紫红色的双溴单体dpp-c6-si7。
5、如图3所示,本实施例中聚合物ptdpptt-c6-si7(图3中m=6,p1=6)的合成方法如下:
在100ml反应瓶中加入(0.089mmol)双溴单体dpp-c6-si7和0.089mmol并噻吩双锡单体,再加入10ml氯苯,用氮气置换40分钟,再加入0.0045mmol催化剂三(二亚苄基丙酮)二钯,以三(邻甲基苯基)磷0.0045mmol为配体,于130℃反应72小时,将反应冷却至室温,加入70ml甲醇沉淀,过滤固体,固体用丙酮索氏提取12小时,再用正己烷提取,旋干得到深紫色的聚合物ptdpptt-c6-si7。
实施例3:合成聚合物ptdppse-c7-si5(m=7,p2=2,主链结构为单硒吩,调整主链和侧链结构是为了得到最优组合)。
如图2所示,本实施例中噻吩dpp单体的溴化合成方法如下:
在100ml反应瓶中加入0.38mmol未溴化的dpp单体,加入15ml的氯仿溶解,加入0.1g自由基捕获剂(tempo),然后将nbs(0.8mmol)溶于氯仿中,逐滴加入到反应瓶中,20min内滴加完成,将反应瓶移至室温,过夜反应;将反应后的产物加水萃取2次,加入无水硫酸镁干燥,过滤后得到得到紫红色的油状物溴化单体;将产物进行硅胶色谱,用石油醚/乙酸乙酯(100:1)作为洗脱液,得到紫红色的双溴单体dpp-c7-si5。
如图3所示,本实施例中聚合物ptdppse-c7-si5(图3中m=7,p2=2)的合成方法如下:
在100ml反应瓶中加入(0.089mmol)双溴单体dpp-c7-si5和0.089mmol单硒吩双锡单体,再加入10ml氯苯,用氮气置换40分钟,再加入0.0045mmol催化剂三(二亚苄基丙酮)二钯,以三(邻甲基苯基)磷0.0045mmol为配体,于130℃反应72小时,将反应冷却至室温,加入70ml甲醇沉淀,过滤固体,固体用丙酮索氏提取12小时,再用氯仿提取,旋干得到深紫色的聚合物ptdppse-c7-si5。
下表为聚合物结构表:
将半导体共轭聚合物溶解在氯仿中,浓度为5mg/ml,使用滴水转膜法将薄膜转移到pdms上,另外准备ps旋涂在绝缘层二氧化硅上形成修饰层。将拉伸后的聚合物薄膜直接转移到修饰层上,最后在聚合物薄膜上蒸镀进电极作为源、漏电极,形成底栅顶接触的晶体管结构,测试其输出特性曲线和转移特性曲线。
图4是对ptdpptt-c6-si5聚合物薄膜进行不同比例拉伸后的电学性能曲线。结果表明,这种可应变的非晶区域可以有效耗散应力,聚合物薄膜在拉伸后电学性能降低的很少,但由于硅链长度有限,效果不明显。
图5是对ptdpptt-c6-si7聚合物薄膜进行不同比例拉伸后的电学性能曲线。结果表明,这种可应变的非晶区域可以更有效耗散应力,聚合物薄膜在拉伸后电学性能降低的很少,计算后,在垂直方向上,100%拉伸下的薄膜仍然能保持初始80%的迁移率,说明延长硅链是一个材料延展性逐渐优化的过程,但延展性和电学性能优化是有范围的。
图6是对ptdpptt-c6-si5,ptdpptt-c6-si7聚合物薄膜拉伸后的高分辨率的afm高度图,ptdpptt-c6-si5在30%应变情况下,高分辨率的afm已经可以看到些许裂纹,而ptdpptt-c6-si7在50%拉伸下仍然没有明显的裂纹。
图7是对ptdpptt-c6-si7材料使用更直接的水面成膜后拉伸的应力-应变曲线,表明其拥有较低的模量108±21mpa。
图8是将聚合物主链改变为更柔软的单硒吩,侧链变为更大体积的分叉有机硅氧烷侧链后,ptdppse-c7-si5聚合物薄膜在拉伸后具有更高的延展性,不同拉伸比例下的薄膜制备的器件电学性能更高,拉伸后电学性能保持的更稳定,表明利用该种结构组合的半导体共轭聚合物具有更优异的本征可拉伸性。
综上所述,本发明指导合成的半导体共轭聚合物利用可调节的烷基直链和有机硅氧烷基团作为侧链,构筑可变形的非晶区域实现拉伸过程中的应力耗散,共轭骨架结构可以多样化,得到兼具高延展性和电荷传输特性的聚合物,可作为空穴型电荷传输材料用于柔性电子器件中,对设计合成高延展性的高性能半导体共轭聚合物具有重要的指导意义。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种石墨烯电极的制备方法
- 电掺杂有机半导体材料和包含它的有机发光器件的制造方法与工艺
- 一类高荧光量子效率芴基给受体H型分子材料及其制备方法和应用与制造工艺
- 聚多巴胺修饰n型半导体材料在构建光电免疫传感器中的应用的制造方法与工艺
- 高分子化合物和使用该高分子化合物的有机半导体元件的制造方法与工艺
- EDOT官能化的共轭聚合物和包含其的光电探测器的制造方法与工艺
- 扩展的非线性并苯衍生物和它们作为有机半导体的用途的制造方法与工艺
- 苉衍生物、光电转换材料及光电转换元件的制造方法与工艺
- 基于二苯并呋喃基团n‑型热激发延迟荧光芳香膦氧主体材料、合成方法及其应用与制造工艺
- 一种4‑溴螺环[芴‑9,9’‑氧杂蒽]的合成方法与制造工艺