一种苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物及其制备方法与催化应用
1.本发明涉及催化剂合成以及聚合物制备技术领域,具体涉及一种苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物及其制备方法与催化应用。
背景技术:
2.环状聚丙交酯具有大环结构且没有链末端,在拓扑结构上与相应的链状聚酯完全不同,导致环状聚丙交酯在物理化学性质上表现出更快的微晶成核、更高的结晶度、更高的热稳定性和更低的特性粘度(bielawski,c.w.;benitez,d.;grubbs,r.h.science 2002,297,2041.)。性质上的巨大差异使环状聚丙交酯在催化、药物载体、粘合剂、材料等领域具有极大的潜在应用价值。目前合成含有环状结构的聚合物虽取得一定的进展,但合成具有高度立体选择性的环状聚合物仍然是一个挑战。
3.2012年,weil,j.等报道含烷氧基侧臂的双芳氧基铝可以有效地通过配位-插入机理催化外消旋丙交酯开环聚合,在2h内获得质均分子量mw为38.82kg/mol和窄分散性的环状聚丙交酯,但该体系催化反应温度高达130℃且转化率未超过80%(weil,j.;mathers,r.t.;getzler,y.d.y.l.macromolecules 2012,45,1118.)。
4.2013年,bourissou,d.采用zn(c6f5)2合成了外消旋丙交酯和ε-己内酯的环状嵌段聚合物,在65℃下可获得分子量达51kg/mol的聚合物,该体系活性低,且需采用特定反应溶剂甲基-四氢呋喃,若采用四氢呋喃会发生开环聚合(piedra-arroni,e.;ladaviere,c.;amgoune,a.;bourissou,d.j.am.chem.soc.2013,135,13306.)。
5.2019年,capacchione,c.合成了一种基于[osso]型配体的铁氯催化剂,以环氧环己烷为溶剂,在80℃下,仅需1h就可以催化400当量外消旋丙交酯聚合,得到环状聚丙交酯,单体转化率72%,分子量为7.9kg/mol,当单体当量增加至10000时仍然可以保持较好的催化活性,反应52小时可以实现97%的单体转化率,分子量达8.5kg/mol,分子量分布(impemba,s.;della,monica,f.;grassi,a.;capacchione,c.;milione,s.chemsuschem 2020,13,141.)。
[0006]
2021年,williams,c.k.采用ce(iii)-nhc引发剂催化外消旋丙交酯聚合,所得环状聚丙交酯的选择性能达到95%,但是聚合物的杂同度(pr)仅0.58(kerr,r.w.f.;ewing,p.m.d.a.;raman,s.k.;smith,a.d.;williams,c.k.;arnold,p.l.acs catal.2021,11,1563.)。
[0007]
2022年,mehrkhodavandi,p.采用阳离子烷基铟催化剂催化外消旋丙交酯扩环聚合,可以获得分子量高达416kg/mol的环状聚合物,但是反应温度需要100℃(goonesinghe,c.;jung,h.-j.;roshandel,h.;diaz,c.;baalbaki,h.a.;nyamayaro,k.;ezhova,m.;hosseini,k.;mehrkhodavandi,p.acs catal.2022,12,7677.)。
[0008]
现有制备环状聚丙交酯的方法存在催化剂活性低,反应需要高温(100℃以上),伴随链状聚合物生成,聚合物的立体选择性控制不佳等问题。因此,亟需一种催化活性高、能
够在温和条件下催化外消旋丙交酯开环聚合的催化剂,可催化制备高分子量、高立体选择性的环状聚丙交酯。
技术实现要素:
[0009]
本发明要解决的技术问题是提供一种苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物及其制备方法与催化应用,本发明提供了一类苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物,在25~80℃下可催化外消旋丙交酯聚合,合成环状杂同聚丙交酯,得到的聚合物中环状结构的聚合物占比>99%,杂同度可高达0.99,表现出高催化活性以及高立体选择性。
[0010]
为了解决上述技术问题,本发明提供以下技术方案:
[0011]
本发明第一方面提供了一种苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物,具有式(i)所示的结构通式:
[0012][0013]
其中,r为甲基,re为nd、yb或y;
[0014]
thf为四氢呋喃。
[0015]
本发明第二方面提供了一种第一方面所述的苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物的制备方法,包括以下步骤:
[0016]
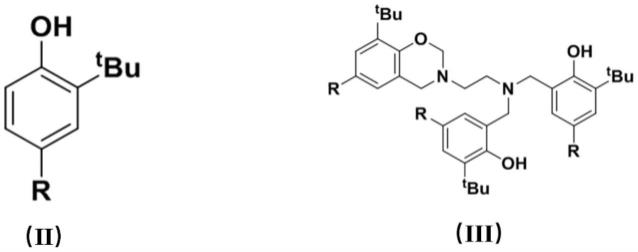
(1)将乙二胺、甲醛与式(ii)所示的取代苯酚在溶剂存在下回流反应,得到式(iii)所示的苯并噁嗪功能化氨基桥联双酚;
[0017]
(2)在无水无氧条件下,将苯并噁嗪功能化氨基桥联双酚与[n(sime3)2]3re(μ-cl)li(thf)3在溶剂存在下反应,得到所述苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物;re为nd、yb或y,thf为四氢呋喃;
[0018]
所述式(ii)、式(iii)的结构如下:
[0019][0020]
其中,r为甲基;
[0021]
进一步地,步骤(1)中,所述回流反应的温度为60~90℃,回流反应的时间为5~6
天。
[0022]
进一步地,步骤(1)中,所述溶剂为乙醇,回流反应的温度优选为78℃,回流反应的时间优选为5天。
[0023]
进一步地,步骤(2)中,所述溶剂为甲苯、四氢呋喃、二氯甲烷中的一种或多种,更优选为甲苯。
[0024]
进一步地,步骤(2)中,所述反应的温度为60~70℃,反应的时间为12~24h。
[0025]
进一步地,步骤(2)中,所述反应的温度优选为65℃,反应的时间优选为12h。
[0026]
本发明第三方面提供了一种第一方面所述的苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物作为催化剂在催化合成环状杂同聚丙交酯中的应用。
[0027]
进一步地,在无水无氧条件下,将外消旋丙交酯与催化剂在第一溶剂存在下,进行搅拌反应,反应结束后加入第二溶剂沉降,过滤得到环状杂同聚丙交酯。
[0028]
进一步地,在高纯氮气保护的手套箱里,向经过脱水脱氧处理的反应瓶中加入稀土金属配合物,然后加入第一溶剂,得到催化剂溶液;将外消旋丙交酯溶于第一溶剂中,得到单体溶液;将催化剂溶液和单体溶液混合,进行搅拌反应。
[0029]
进一步地,所述催化剂为苯并噁嗪功能化氨基桥联多芳氧基镱金属配合物,即所述苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物中稀土金属为镱。
[0030]
进一步地,所述第一溶剂为甲苯、四氢呋喃、二氯甲烷中的一种或多种,更优选为四氢呋喃。
[0031]
进一步地,在单体溶液与催化剂溶液混合后形成的混合溶液中,所述单体的浓度为2.0~6.0mol/l,更优选为2.0~4.0mol/l。
[0032]
进一步地,所述外消旋丙交酯与催化剂的摩尔比为50~2000:1,更优选为200~400:1。
[0033]
进一步地,所述反应的温度25~80℃,更优选为25~40℃。
[0034]
进一步地,所述反应的时间为0.5~72h,更优选为2~10h。
[0035]
进一步地,反应结束后加入第二溶剂沉降,过滤后使用第二溶剂洗涤2~3次,真空干燥得到环状杂同聚丙交酯。
[0036]
进一步地,沉降及洗涤使用的第二溶剂为95%乙醇水溶液或正己烷。
[0037]
进一步地,所述环状杂同聚丙交酯的杂同度(pr)为0.69~0.99,优选杂同度(pr)为0.80~0.99。
[0038]
本发明的有益效果:
[0039]
1.本发明提供了一类苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物,这类多芳氧基稀土金属配合物的制备方法简单,且制备得到的离子半径小的稀土金属配合物可作为催化剂用于催化合成高杂同度的环状杂同聚丙交酯;另外,本发明制备的稀土金属配合物可通过调节稀土配合物的空间结构来调控催化剂的活性和立体选择性。
[0040]
2.本发明采用苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物作为催化剂,具有高催化活性,可室温催化外消旋丙交酯聚合,合成环状杂同聚丙交酯,该合成反应的反应条件温和且聚合物产率高(高达99%),产物中环状结构的聚合物占比大于99%,环状聚丙交酯的合成效率高且产物易于分离提纯;由本发明所述苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物作为催化剂催化合成的环状杂同聚丙交酯的杂同度高达0.99,表现出高立
体选择性,可高效合成数均分子量为10~100kg/mol的环状杂同聚丙交酯。
附图说明
[0041]
图1为实施例2~4所述苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物的合成示意图;
[0042]
图2为实施例3所制备的配合物l
1-nr2
y的核磁氢谱图;
[0043]
图3为实施例3所制备的配合物l
1-nr2
y的核磁碳谱图;
[0044]
图4为实施例5所制备的环状聚(外消旋丙交酯)的基质辅助激光解吸电离飞行时间(maldi-tof)质谱图;
[0045]
图5为实施例7所得聚合物进行核磁共振1h nmr同核去耦分析图谱。
具体实施方式
[0046]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0047]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0048]
实施例1苯并噁嗪功能化氨基桥联双酚h2l1合成:
[0049]
以乙二胺(3.00g,50mmol)、甲醛(24.30g,300mmol)、2-叔丁基-4-甲基苯酚(32.85g,200mmol)为原料,在78℃下乙醇中回流搅拌5天,生成大量固体,减压抽滤并用乙醇洗涤,得到白色固体粉末23.13g,产率77%。干燥后的固体装入瓶中,无水无氧处理后,封瓶备用。反应式如下:
[0050][0051]
元素分析(%):c,77.72;h,9.80;n,4.83。
[0052]
核磁共振1h谱(400mhz,cdcl3,25℃):δ8.99(s,2h,oh),7.01(s,2h,arh),6.91(s,1h,arh),6.73(s,2h,arh),6.52(s,1h,arh),4.92(s,2h,aroch2),4.03(s,2h,arch2),3.62(s,4h,arch2),3.02(t,j=5.7hz,2h,nch2ch2n),2.66(t,j=5.7hz,2h,nch2ch2n),2.24(s,6h,arch3),2.22(s,3h,arch3),1.41(s,18h,c(ch3)3),1.33(s,9h,c(ch3)3)。
[0053]
核磁共振
13
c谱(101mhz,cdcl3):δ151.9,148.9,136.3,135.7,128.2,127.7,126.3,126.1,124.7,124.6,121.1,117.6,80.1,55.2,48.0,47.9,46.5,33.7,33.5,28.5,19.8,19.7。
[0054]
实施例2苯并噁嗪功能化氨基桥联多芳氧基钕金属配合物l
1-nr2
nd合成:
[0055]
在无水无氧条件下,称取1.15g(1.3mmol)的[n(sime
3)2
]3nd(μ-cl)li(thf)3和
0.78g(1.3mmol)的h2l1于反应瓶中,加入约20ml的甲苯,反应液澄清透明,封瓶。将反应瓶置于60℃油浴中,搅拌反应1天,反应结束后,离心,减压浓缩上层清液,加入约2ml四氢呋喃和4ml正己烷的混合溶液重结晶,析出蓝色块状晶体,用正己烷洗涤晶体,减压干燥得蓝色固体粉末0.87g,产率69%。得到苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物l
1-nr2
nd,反应式如图1。
[0056]
元素分析(%):c,59.84;h,8.76;n,4.29。
[0057]
红外光谱(kbr,cm-1
):2951(s),2904(s),1605(s),1466(s),1413(s),1382(s),1349(s),1299(s),1121(s),1090(s),1021(s),943(s),912(s),842(vs),776(s),622(s)。
[0058]
实施例3苯并噁嗪功能化氨基桥联多芳氧基钇金属配合物l
1-nr2
y合成:
[0059]
在无水无氧条件下,称取1.08g(1.3mmol)的[n(sime
3)2
]3y(μ-cl)li(thf)3和0.78g(1.3mmol)的h2l1于反应瓶中,加入约20ml的甲苯,反应液澄清透明,封瓶。将反应瓶置于60℃油浴中,搅拌反应1天,反应结束后,离心,减压浓缩上层清液,加入约2ml四氢呋喃和6ml正己烷的混合溶液重结晶,析出白色块状晶体,用正己烷洗涤晶体,减压干燥得白色固体粉末0.92g,产率77%。得到苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物l
1-nr2
y,反应式如图1。
[0060]
元素分析(%):c,63.96;h,8.55;n,4.02。
[0061]
红外光谱(kbr,cm-1
):2947(s),2866(s),1463(s),1437(s),1414(s),1383(s),1300(s),1266(s),1123(s),1093(s),1020(s),913(s),844(vs),781(s),622(s)。
[0062]
核磁共振1h谱(400mhz,thf-d8,25℃):δ7.30(d,j=1.9hz,1h,arh),7.27(d,j=2.0hz,1h,arh),7.18(d,j=2.0hz,1h,arh),6.92(s,1h,arh),6.79-6.75(m,1h,arh),6.54(s,1h,arh),4.05(d,j=12.7hz,1h,arch2),3.95(d,j=11.6hz,1h,arch2),3.73(d,j=14.3hz,1h,arch2),3.69(s,2h,arch2),3.09(t,j=11.4hz,1h,arch2),2.83(s,1h,arch2),2.75-2.65(m,1h,arch2),2.61(d,j=12.5hz,1h,arch2),2.45(d,j=14.1hz,1h,arch2),2.37(s,3h,ch3),2.33(s,3h,ch3),2.28(s,3h,ch3),1.82(d,j=16.9hz,1h,arch2),1.78(s,1h,arch2),1.68(s,9h,c(ch3)3),1.64(s,9h,c(ch3)3),1.60(s,9h,c(ch3)3),0.06(s,18h,si(ch3)3)(图2)。
[0063]
核磁共振
13
c谱(101mhz,thf-d8):δ161.5,137.2,136.4,130.6,129.5,129.0,128.3,126.3,125.1,123.1,122.9,69.2,64.7,63.4,60.7,57.0,54.9,48.2,35.4,35.3,30.6,30.5,30.4,26.4,25.8,25.6,25.4,21.0,3.7,2.6(图3)。
[0064]
实施例4苯并噁嗪功能化氨基桥联多芳氧基镱金属配合物l
1-nr2
yb合成:
[0065]
在无水无氧条件下,称取1.18g(1.3mmol)的[n(sime
3)2
]3yb(μ-cl)li(thf)3和0.78g(1.3mmol)的h2l2于反应瓶中,加入约20ml的甲苯,反应液澄清透明,封瓶。将反应瓶置于60℃油浴中,搅拌反应1天,反应结束后,离心,减压浓缩上层清液,加入约2ml四氢呋喃和4ml正己烷的混合溶液重结晶,析出块状晶体,用正己烷洗涤晶体,减压干燥得固体粉末0.94g,产率72%。得到苯并噁嗪功能化氨基桥联多芳氧基稀土金属配合物l
1-nr2
yb,反应式如图1。
[0066]
元素分析(%):c,58.89;h,8.71;n,4.15。
[0067]
红外光谱(kbr,cm-1
):2950(s),2908(s),1467(s),1414(s),1373(s),1314(s),1299(s),1252(s),1150(s),1039(s),913(s),841(vs),778(s),621(s)。
[0068]
实施例5l
1-nr2
nd配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0069]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.009g l
1-nr2
nd配合物(0.009mmol),再加入0.5ml四氢呋喃,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.22g,产率85%)。
[0070]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=19.5kg/mol,分子量分布通过基质辅助激光解吸电离飞行时间(maldi-tof)质谱对聚合物进行表征(图4),显示所得聚合物均具有环状拓扑结构。对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.93。
[0071]
实施例6l
1-nr2
y配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0072]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.009g l
1-nr2
y配合物(0.009mmol),再加入0.5ml四氢呋喃,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.17g,产率63%)。
[0073]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=19.2kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.94。
[0074]
实施例7l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0075]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.010g l
1-nr2
yb配合物(0.01mmol),再加入0.5ml四氢呋喃,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.12g,产率44%)。
[0076]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=13.5kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,结果如图5所示,计算得到聚合物的杂同度(pr)为0.99。
[0077]
实施例8l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:400)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0078]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.017g l
1-nr2
yb配合物(0.017mmol),再加入1.0ml四氢呋喃,得到催化剂的溶液;将1.00g外消旋丙交酯(6.94mmol)溶于2.5ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应16h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.98g,产率98%)。
[0079]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=89.93kg/
mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.98。
[0080]
实施例9l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:2000)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0081]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.003g l
1-nr2
yb配合物(0.003mmol),再加入1.0ml四氢呋喃,得到催化剂的溶液;将1.00g外消旋丙交酯(6.94mmol)溶于2.5ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应72h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.96g,产率96%)。
[0082]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=91.0kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.98。
[0083]
实施例10l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0084]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.010g l
1-nr2
yb配合物(0.01mmol),再加入0.5ml二氯甲烷,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml二氯甲烷中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.08g,产率30%)。
[0085]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=10.4kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.69。
[0086]
实施例11l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0087]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.010g l
1-nr2
yb配合物(0.01mmol),再加入0.5ml甲苯,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml甲苯中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.09g,产率35%)。
[0088]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=11.8kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.84。
[0089]
实施例12l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0090]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.010g l
1-nr2
yb配合物(0.01mmol),再加入0.5ml四氢呋喃,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于40℃下搅拌反应4h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3
次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.17g,产率64%)。
[0091]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=19.7kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.97。
[0092]
实施例13l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:200)开环聚合,生成环状聚合物([rac-la]=2.0mol/l)
[0093]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.010g l
1-nr2
yb配合物(0.01mmol),再加入0.5ml四氢呋喃,得到催化剂的溶液;将0.27g外消旋丙交酯(1.88mmol)溶于0.45ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应10h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.26g,产率98%)。
[0094]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=62.3kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.92。
[0095]
实施例14l
1-nr2
yb配合物催化外消旋丙交酯(摩尔比1:400)开环聚合,生成环状聚合物([rac-la]=4.0mol/l)
[0096]
在高纯氮气保护的手套箱里,向经过脱水脱氧处理过的10ml反应瓶中加入0.047g l
1-nr2
yb配合物(0.047mmol),再加入1.0ml四氢呋喃,得到催化剂的溶液;将2.7g外消旋丙交酯(18.7mmol)溶于3.7ml四氢呋喃中,得到单体的溶液。将催化剂溶液和单体溶液混合,于25℃下搅拌反应12h。将反应瓶转出手套箱,使用正己烷沉降聚合物。过滤,用正己烷淋洗2-3次。所得聚合物置于真空干燥箱中干燥至恒重,得到聚合产物(0.99g,产率99%)。
[0097]
对所得聚合物进行凝胶渗透色谱(gpc)分析,测得聚合物的分子量mn=91.01kg/mol,分子量分布对所得聚合物进行核磁共振1h nmr同核去耦分析,计算得到聚合物的杂同度(pr)为0.98。
[0098]
以上所述实施例仅是为充分说明本发明而所举的较佳的施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1