近红外磷光发射材料及其制备方法和应用
本发明涉及磷光材料技术领域,尤其是涉及一种近红外磷光发射材料及其制备方法和应用。
背景技术:
纯有机室温磷光(rtp)材料作为传统无机/金属有机荧光粉的替代品,引起了人们越来越多的关注。作为独特的延迟发光可以广泛的应用于急救灯、防伪和生物成像等领域。但是具有长寿命高效率的纯有机磷光发射仍面临严峻的挑战,主要是因为从单线态通过自旋轨道耦合发生系间穿越到三线态是自旋禁阻,并且三线态非常容易受到环境中的水和空气影响而发生非辐射跃迁失活。更有甚者,纯有机材料的磷光的发射难以在红光或者近红外区域,因此对于发展长寿命高效率发波长较长的材料仍具有重大的挑战。
现有技术中,为了使有机物产生室温磷光,通常在有机物中引入重金属原子,利用重金属的重原子效应诱导有机物三线态的产生,以促进有机物室温磷光的产生。然而,重金属成本高,且环保性差,急需研发不含重金属原子且具有优异磷光性能且发射波长在近红外区域的有机室温磷光材料。
有鉴于此,特提出本发明。
技术实现要素:
本发明的第一目的在于提供近红外磷光发射材料,具有室温纯磷光发射性能且发射波长在近红外区。
本发明的第二目的在于提供近红外磷光发射材料的制备方法。
本发明的第三目的在于提供近红外磷光发射材料的应用。
为了实现本发明的上述目的,特采用以下技术方案:
近红外磷光发射材料,包括主体化合物和客体化合物;所述客体化合物结构式如下:
在本发明的具体实施方式中,所述烷氧基包括甲氧基和乙氧基中的至少一种。进一步的,所述烷氧基为甲氧基。
在本发明的具体实施方式中,所述卤素原子包括f、cl、br和i中的至少一种。进一步的,所述卤素原子为f。
在本发明的具体实施方式中,所述胺基为
在本发明的具体实施方式中,所述客体化合物包括如下结构中的至少一种:
在本发明的具体实施方式中,所述主体化合物与所述客体化合物的摩尔比为(50~100000)﹕1,优选为(50~10000)﹕1,更优选为500﹕1。
在本发明的具体实施方式中,所述主体化合物包括二苯甲酮。
在本发明的具体实施方式中,所述近红外磷光发射材料的激发波长为320~420nm,优选为365~400nm。
在本发明的具体实施方式中,所述近红外磷光发射材料的发射波长为590~750nm。
在本发明的具体实施方式中,所述近红外磷光发射材料的量子产量≥4.8%,如为4.8%~9.6%。
本发明还提供了上述任意一种所述近红外磷光发射材料的制备方法,包括如下步骤:
主体化合物和客体化合物于加热条件下熔融混合后,冷却。
在本发明的具体实施方式中,还包括:将所述冷却后的物料制成粉状。在实际操作中,可通过研磨的方式制成粉状。
在本发明的具体实施方式中,所述加热的温度为50~60℃。
在本发明的具体实施方式中,所述客体化合物的制备方法包括:
在非氧化性气体保护下,化合物a与化合物b在催化剂和无机碱的作用下,于溶剂中在80~85℃条件下进行偶联反应;
其中,所述化合物a和所述化合物b的结构式分别如下:
在本发明的具体实施方式中,所述催化剂为四(三苯基膦)钯。
在本发明的具体实施方式中,所述无机碱包括碳酸钾。
在本发明的具体实施方式中,所述化合物a与所述化合物b的摩尔比1﹕(2~3)。
在本发明的具体实施方式中,所述溶剂包括水和四氢呋喃。进一步的,所述水和四氢呋喃的体积比为1﹕(8~12),优选为1﹕10。
在本发明的具体实施方式中,所述偶联反应的时间为24~48h。
在本发明的具体实施方式中,所述客体化合物的制备还包括后处理;所述后处理包括:将所述偶联反应后的物料冷却至室温后,过滤收集滤液;除去所述滤液中的溶剂得到粗产物,通过柱色谱法对所述粗产物进行分离纯化。
本发明还提供了所述近红外磷光发射材料在制备用于生物检测或细胞成像的试剂或试剂盒中的应用。
本发明还提供了所述近红外磷光发射材料在信息储存或防伪中的应用。
与现有技术相比,本发明的有益效果为:
(1)本发明的磷光发射材料表现出有机室温纯磷光发射,并且量子产量在5%左右;
(2)本发明的磷光发射材料在可见光的激发波长下,可以发射出红色或近红外的磷光,可以用于生物检测或细胞成像,以及信息储存或防伪等领域。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
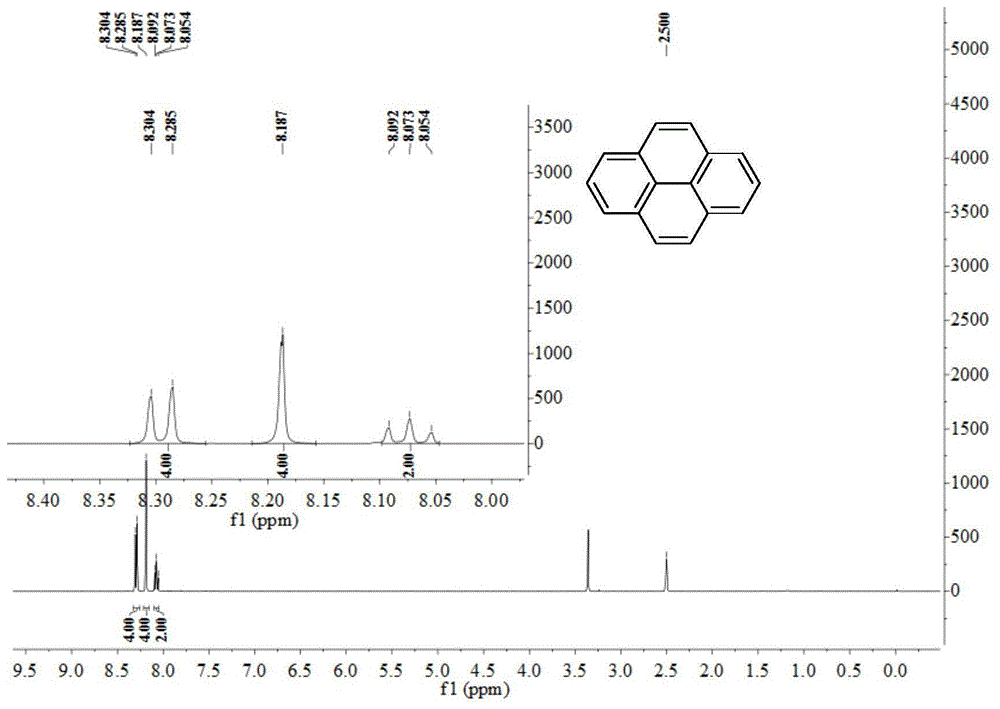
图1为本发明实施例提供的客体化合物py的核磁共振氢谱;
图2为本发明实施例提供的客体化合物py的核磁共振碳谱;
图3为本发明实施例提供的客体化合物fpy的核磁共振氢谱;
图4为本发明实施例提供的客体化合物fpy的核磁共振碳谱;
图5为本发明实施例提供的客体化合物mopy的核磁共振氢谱;
图6为本发明实施例提供的客体化合物mopy的核磁共振碳谱;
图7为本发明实施例提供的客体化合物mapy的核磁共振氢谱;
图8为本发明实施例提供的客体化合物mapy的核磁共振碳谱;
图9为本发明实施例提供的客体化合物dmopy的核磁共振氢谱;
图10为本发明实施例提供的客体化合物dmapy的核磁共振氢谱;
图11为本发明实施例提供的客体化合物py、fpy、mopy、mapy、dmopy和dmapy在360nm激发波长下的溶液和固态下的荧光光谱图;
图12为本发明实施例提供的客体化合物py、fpy、mopy、mapy、dmopy和dmapy在固态和溶液态下的365nm紫外光照下照片;
图13为本发明实施例提供的固态客体化合物py、fpy、mopy、mapy、dmopy和dmapy的稳态延时衰减曲线;
图14为本发明实施例提供的py与二苯甲酮在不同掺杂浓度下得到的发射材料的稳态和瞬态光谱;
图15为本发明实施例提供的py与二苯甲酮在不同掺杂浓度下得到的发射材料的荧光磷光照片;
图16为本发明实施例提供的不同掺杂的磷光发射材料的瞬态光谱图和稳态光谱图;
图17为本发明实施例提供的不同掺杂的磷光发射材料的紫外光照前后的发光颜色变化;
图18为本发明实施例提供的不同掺杂的磷光发射材料py/bpo-3、fpy/bpo、mopy/bpo、mapy/bpo、dmopy/bpo、dmapy/bpo的cie坐标;
图19为本发明实施例提供的不同掺杂的磷光发射材料的时间延迟衰减曲线;
图20为本发明实施例提供的不同掺杂的磷光发射材料的磷光延迟光谱(650nm);
图21为本发明实施例提供的不同掺杂的磷光发射材料的磷光延迟光谱(700nm)。
具体实施方式
下面将结合附图和具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
本发明的结构式中所涉及的“*”,表示的是含有“*”的结构式的基团通过“*”的位置与化合物其余部分连接。
近红外磷光发射材料,包括主体化合物和客体化合物;所述客体化合物结构式如下:
在本发明的具体实施方式中,所述烷氧基包括甲氧基和乙氧基中的至少一种。进一步的,所述烷氧基为甲氧基。
在本发明的具体实施方式中,所述卤素原子包括f、cl、br和i中的至少一种。进一步的,所述卤素原子为f。
在本发明的具体实施方式中,所述胺基为
在本发明的具体实施方式中,所述客体化合物包括如下结构中的至少一种:
在本发明的具体实施方式中,所述主体化合物与所述客体化合物的摩尔比为(50~100000)﹕1,优选为(50~10000)﹕1,更优选为(100~2000),进一步优选为(500~1500)﹕1,如500﹕1。
如在不同实施方式中,所述主体化合物与所述客体化合物的摩尔比可以为50﹕1、100﹕1、150﹕1、200﹕1、300﹕1、400﹕1、500﹕1、600﹕1、700﹕1、800﹕1、900﹕1、1000﹕1、2000﹕1、3000﹕1、4000﹕1、5000﹕1、6000﹕1、7000﹕1、8000﹕1、9000﹕1、10000﹕1、20000﹕1、30000﹕1、40000﹕1、50000﹕1、60000﹕1、70000﹕1、80000﹕1、90000﹕1、100000﹕1等等。
采用上述掺杂比例得到的发射材料,均具有一定强度的磷光。
在本发明的具体实施方式中,所述主体化合物包括二苯甲酮。
二苯甲酮具有熔点低、结晶度好、能发出磷光等特点。采用二苯甲酮作为主体,其低熔点能够有助于材料的加工,良好的结晶度有利于为客体分子提供刚性的环境。
在本发明的具体实施方式中,所述近红外磷光发射材料的激发波长为320~420nm,优选为365~400nm。
采用上述激发波长,所述磷光发射材料发射近红外磷光。在320~420nm范围内激发,如在380nm激发可以发射出近红外的室温磷光。
本发明所述的磷光发射材料具有红色或近红外室温磷光。
在本发明的具体实施方式中,所述近红外磷光发射材料的发射波长为590~750nm。
在本发明的具体实施方式中,所述近红外磷光发射材料的量子产量≥4.8%,如为4.8%~9.6%。
本发明还提供了上述任意一种所述近红外磷光发射材料的制备方法,包括如下步骤:
主体化合物和客体化合物于加热条件下熔融混合后,冷却。
在本发明的具体实施方式中,还包括:将所述冷却后的物料制成粉状。在实际操作中,可通过研磨的方式制成粉状。在实际操作中,可采用自然冷却方式,在冷却过程中会析出晶状材料,即为所述磷光发射材料。
在本发明的具体实施方式中,所述加热的温度为50~60℃。加热温度可根据实际需求进行调整,使所述主体化合物与所述客体化合物熔融混匀即可,比如使所述客体化合物熔融,并能够使所述主体化合物溶解在客体化合物的熔融物中。
在本发明的具体实施方式中,所述客体化合物的制备方法包括:
在非氧化性气体保护下,化合物a与化合物b在催化剂和无机碱的作用下,于溶剂中在80~85℃条件下进行偶联反应;
其中,所述化合物a和所述化合物b的结构式分别如下:
在实际操作中,所述非氧化性气体包括氮气和/或氩气。
在本发明的具体实施方式中,所述催化剂为四(三苯基膦)钯。所述催化剂的用量为常规催化剂用量,如四(三苯基膦)钯的摩尔量为所述化合物b的摩尔量的3%~5%,如5%。
在本发明的具体实施方式中,所述无机碱包括碳酸钾。所述碱的用量为所述化合物b的摩尔量的3%~5%,如5%。
在本发明的具体实施方式中,所述化合物a与所述化合物b的摩尔比1﹕(2~3)。
在本发明的具体实施方式中,所述溶剂包括水和四氢呋喃。进一步的,所述水和四氢呋喃的体积比为1﹕(8~12),优选为1﹕10。
在本发明的具体实施方式中,所述偶联反应的时间为24~48h。
本发明的客体化合物的合成路线可以如下:
在本发明的具体实施方式中,所述客体化合物的制备还包括后处理;所述后处理包括:将所述偶联反应后的物料冷却至室温后,过滤收集滤液;除去所述滤液中的溶剂得到粗产物,通过柱色谱法对所述粗产物进行分离纯化。
本发明还提供了所述近红外磷光发射材料在制备用于生物检测或细胞成像的试剂或试剂盒中的应用。
本发明还提供了所述近红外磷光发射材料在信息储存或防伪中的应用。
实施例1
本实施例提供了磷光发射材料的制备方法,包括如下步骤:
按比例称取客体化合物和主体化合物二苯甲酮,然后混合,再加热至50~60℃,使主体化合物二苯甲酮熔融并将客体化合物溶解,二者混合均匀后,自然冷却至室温,得到磷光发射材料。
其中,磷光发射材料编号、客体化合物种类、客体化合物与主体化合物的比例等信息见表1。
表1不同磷光发射材料的原料信息等
其中,客体py直接购买(安耐吉化学)得到,其它客体化合物fpy、mopy、mapy、dmopy和dmapy的合成路线分别如下:
客体化合物fpy的制备过程包括:称取对氟苯硼酸(20mmol)、1-溴芘(10mmol)、pd(pph3)4(1.0mmol)、碳酸钾(1.0mmol)、h2o(1.0ml)、四氢呋喃(10.0ml),置于反应容器中,在氮气氛围中于80℃反应24h。反应结束后冷却至室温,然后将反应后的物料过滤以除去催化剂等,收集过滤后的滤液;然后减压除去滤液中的溶剂得到粗产物,将所述粗产物通过常规柱色谱法(洗脱剂为石油醚和乙酸乙酯,二者体积比为100﹕1)分离纯化得到纯的客体化合物fpy。
客体化合物mapy的制备过程参考fpy,区别在于,将对氟苯硼酸替换为等摩尔的4-(n,n-二甲氨基)苯硼酸。
客体化合物mopy的制备过程参考fpy,区别在于,将对氟苯硼酸替换为等摩尔的对甲氧基苯硼酸。
客体化合物dmapy的制备过程包括:称取4-(n,n-二甲氨基)苯硼酸(20mmol)、1,6-二溴芘(10mmol)、pd(pph3)4(1.0mmol)、碳酸钾(1.0mmol)、h2o(1.0ml)、四氢呋喃(10.0ml),置于反应容器中,在氮气氛围中于80℃反应24h。反应结束后冷却至室温,然后将反应后的物料过滤以除去催化剂等,收集过滤后的滤液;然后减压除去滤液中的溶剂得到粗产物,将所述粗产物通过常规柱色谱法(洗脱剂为石油醚和乙酸乙酯,二者体积比为50﹕1)分离纯化得到纯的客体化合物dmapy。
客体化合物dmopy的制备过程参考dmapy,区别在于,将4-(n,n-二甲氨基)苯硼酸替换为等摩尔的对甲氧基苯硼酸。
客体化合物py、fpy、mopy、mapy、dmopy和dmapy的核磁氢谱和/或核磁碳谱分别见图1~图10,根据表征结果证实了客体化合物py、fpy、mopy、mapy、dmopy和dmapy的结构。
图11为客体化合物py、fpy、mopy、mapy、dmopy和dmapy在360nm激发波长下的溶液和固态下的荧光光谱图;图12为客体化合物py、fpy、mopy、mapy、dmopy和dmapy在固态和溶液态下的365nm紫外光照下的照片。其中,各个溶液中,客体化合物的浓度为1.0*10-5mol/l,溶剂为thf。
图13为客体化合物py、fpy、mopy、mapy、dmopy和dmapy在固态下的荧光延迟时间衰减谱图。
实验例1
为了对比说明不同磷光发射材料的性质,对实施例1制得的不同磷光发射材料的性能进行表征。其中,将实施例1中的各个发射材料在研磨皿中磨成粉状后进行后续表征。
图14为本发明实施例提供的py与二苯甲酮,在不同掺杂浓度下的发射材料的稳态和瞬态光谱。从图中可知,几种发射材料之间相比,客体化合物与主体化合物的摩尔比为1﹕500的py/bpo-3具有更强的磷光强度。
图15为本发明实施例提供的py与二苯甲酮,在不同掺杂浓度下的发射材料py/bpo的荧光磷光照片。其中,图15中,“turnon”对应的一行是不同掺杂浓度的发射材料在365nm波长紫外光照射下对应的照片;“turnoff”对应的一行是不同掺杂浓度的发射材料在撤去365nm波长紫外光照射后对应的照片。
图16为本发明实施例提供的不同掺杂发射材料的瞬态光谱图和稳态光谱图。
图17为本发明实施例提供的不同掺杂发射材料在365nm波长的紫外光照前后的发光颜色变化。其中,“turnon”对应的一列为不同的发射材料在365nm波长紫外光照射下对应的照片;“turnoff”对应的一列分别为不同的发射材料在撤去365nm波长紫外光照后不同时长对应的照片。
图18为本发明实施例提供的不同掺杂的磷光发射材料py/bpo-3、fpy/bpo、mopy/bpo、mapy/bpo、dmopy/bpo、dmapy/bpo的cie坐标。
图19为本发明实施例提供的不同掺杂发射材料的时间延迟衰减曲线。
图20为本发明实施例提供的不同掺杂发射材料的磷光延迟光谱(650nm)。
图21为本发明实施例提供的不同掺杂发射材料的磷光延迟光谱(700nm)。
本发明得到的磷光发射材料表现出有机室温纯磷光发射的性质,并且当所述主体化合物与所述客体化合物的摩尔比为1﹕500时,py/bpo-3的磷光发射波长为599nm和659nm,量子产率高达9.6%,寿命为324ms和328ms;fpy/bpo的磷光发射波长为623nm和685nm,量子产率高达9.5%,寿命为235ms和255ms;mopy/bpo的磷光发射波长为628nm和690nm,量子产率高达5.3%,寿命为210ms和215ms;mapy/bpo的磷光发射波长为641nm和700nm,量子产率高达8.3%,寿命为199ms和202ms;dmopy/bpo的磷光发射波长为653nm和706nm,量子产率高达5.5%,寿命为173ms和165ms;dmapy/bpo的磷光发射波长为676nm和730nm,量子产率高达4.8%,寿命为106ms和81ms。本发明可以在可见光的激发波长下,即约400nm的激发波长下发射出红色或近红外的室温磷光,因此本发明的发射材料可以作为生物检测,细胞成像等应用。另,本发明的磷光发射材料还可用于信息储存或防伪领域,比如用于制备书写或打印信息的墨水等。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!