一种析氧Fe掺杂二硒化钴@N‑CT复合催化剂及其制备方法和应用与流程
本发明涉及一种析氧(oer)催化剂及其制备和应用方法,特别涉及一种用于电催化水分解的析氧n掺杂纳米碳管包覆fe掺杂cose2纳米颗粒复合催化剂(fe-dopedcose2@n-ct,记为fcs@n-ct)及制备方法,属于电催化技术领域。
背景技术:
能源是现代社会发展的三大支柱之一。随着现代社会的发展,不可再生的化石能源日益枯竭,氢气(h2)作为可再生的新能源,因具有高能量密度、来源广泛和环境友好等优点,获得了研究人员的广泛关注。目前,工业上一般利用天然气或通过水煤气方法来制氢气,电解水因为高能耗而未被采用。然而地球71%以上的表面被水所覆盖,所以以水制氢具有极大的吸引力。水的分解反应为2h2o→2h2+o2,它包含析氢反应(her,2h++2e–→2h2,e0=0vvs.rhe)和析氧反应(oer,2h2o→4h++o2+4e–,e0=1.23vvs.rhe)两个半反应。电解水的效率低且能耗高,其中的一个原因是因为oer反应包含复杂的4电子反应过程并需要形成新的o–o键,从而产生了高的过电位η。所以,在电解水的实际应用中需要高效的oer催化剂来降低反应过程中的η。迄今,最高效的oer催化剂是贵金属ru或ir基的材料,如ruo2和iro2。然而,与pt基氧还原催化剂一样,高成本、稀缺性及低稳定性也是限制它们广泛应用的瓶颈。因此,许多由过渡金属氧化物、过渡金属磷化物、过渡金属硒化物、过渡金属纳米粒子等形成的材料,因具有成本低廉、来源广泛、易于合成等优点,它们的oer性能被研究人员所广泛关注。其中,钴硒化合物作为一种高活性的co基电化学催化剂,因低成本和具有高的热稳定性及化学稳定性等优点而吸引了大量的关注。cose2在理论上具有较高的oer催化性能,但实际使用过程中,由于其导电率低、活性位点少,实际催化性能偏低,应用受到局限。将cose2与碳材料复合,可以控制cose2的形貌并提高复合材料的比表面积,暴露出更多的oer活性位点,从而获得改善的oer催化性能。杂原子掺杂能改变催化剂的电子结构,增加结构和原子的缺陷,从而得到改善的电催化性能。如中国专利(cn105609322a)公开了一种硒化钴/氮掺杂碳复合材料及其制备方法和应用,其以有机金属骨架zif-67为模板制备硒化钴/氮掺杂碳复合材料,硒化钴为一硒化钴或二硒化钴纳米颗粒,均匀地分散在由氮掺杂的碳骨架上。该复合物具有良好的介孔结构和优良的导电性,用作超级电容器的电极材料时,具有高比电容和良好稳定性。但是其制备方法步骤繁琐,且其制备的硒化钴/氮掺杂碳复合材料主要是作为电容器电极材料。
技术实现要素:
针对现有技术中单一cose2作为oer电催化剂存在活性和电导率低的缺陷,本发明的目的之一是在于提供一种催化活性高、稳定性好,综合催化性能接近甚至超过商用贵金属ruo2的fcs@n-ct复合催化剂。
本发明的第二个目的是在于提供一种操作简单、低成本制备所述析氧fcs@n-ct复合催化剂的方法;该方法能使n掺杂纳米碳管和fe掺杂co纳米颗粒一步生成且原位复合,工艺过程简单,满足工业化生产要求。
本发明的第三个目的在于提供所述高性能析氧fcs@n-ct复合催化剂在水分解中的应用,在碱性介质中,析氧fcs@n-ct复合催化剂综合催化性能接近甚至超过商用贵金属ruo2催化剂。
为了实现上述技术目的,本发明提供了一种析氧fcs@n-ct复合催化剂,该复合催化剂由n掺杂纳米碳管包覆fe掺杂cose2纳米颗粒构成。
优选的方案,所述析氧fcs@n-ct复合催化剂由以下质量百分比组分组成:fe掺杂cose2纳米颗粒75%~90%;n掺杂纳米碳管10%~25%;所述fe掺杂cose2纳米颗粒中fe质量百分比含量为10%~20%;所述n掺杂纳米碳管中n质量百分比含量为1%~10%。
较优选的方案,所述析氧fcs@n-ct复合催化剂由以下质量百分比组分组成:fe掺杂cose2纳米颗粒80%~85%;n掺杂纳米碳管15%~20%;所述fe掺杂cose2纳米颗粒中fe质量百分比含量为13%~17%;所述n掺杂纳米碳管中n质量百分比含量为3%~8%。
本发明还提供了一种析氧fcs@n-ct复合催化剂的制备方法,该方法是将含醋酸钴、硝酸铁和尿素的溶液蒸发、烘干后,置于500~1000℃温度下热处理,即得n掺杂纳米碳管包覆fe掺杂co纳米颗粒;所述n掺杂纳米碳管包覆fe掺杂co纳米颗粒与se粉混合后,置于保护气氛下,于300~700℃温度下进行硒化处理,即得。
优选的方案,醋酸钴、硝酸铁和尿素的质量百分比组成为(2.1%~6.9%):(0.3%~1.2%):(91.9%~97.6%)。
优选的方案,所述n掺杂纳米碳管包覆fe掺杂co纳米颗粒与se粉的质量百分比组成为(10%~25%):(75%~90%)。
优选的方案,所述热处理的温度为750~850℃;最佳热处理温度为800℃。
优选的方案,所述硒化处理的温度为450~550℃;最佳硒化处理温度为500℃。
优选的方案,所述热处理的时间为0.5~2h;较优选的热处理时间为0.8~1.2h。
优选的方案,所述硒化处理的时间为1~5h;较优选的硒化处理时间为2~4h。
本发明还提供了一种析氧fcs@n-ct复合催化剂的应用,将其作为析氧催化剂应用于电解水。
优选的方案,在碱性条件下,作为析氧催化剂用于电解水。
相对现有技术,本发明的技术方案带来的有益技术效果:
1、本发明的析氧fcs@n-ct复合催化剂由n掺杂纳米碳管包覆fe掺杂cose2纳米颗粒构成,各组分之间协同增效作用明显,使复合物表现出较高的催化活性。析氧fcs@n-ct复合催化剂利用氮掺杂纳米碳管包覆铁掺杂硒化钴颗粒,增加了铁掺杂硒化钴颗粒的分散稳定性,可以有效增加铁掺杂硒化钴的比表面积以及改善其形貌特征,暴露出更多的oer活性位点,从而使复合催化剂具备更高的oer催化性能。氮掺杂纳米碳管利用其极性氮原子与fe掺杂cose2纳米颗粒中金属配位,大大提高了复合材料的稳定性,同时n掺杂纳米碳管中氮掺杂位点也具有oer催化活性,增加了复合材料的oer催化活性位点。而铁元素掺杂cose2能增加cose2的晶体缺陷,起到加强电化学催化剂的催化性能。因此,由n掺杂纳米碳管包覆fe掺杂cose2纳米颗粒构成的cose2@n-ct复合催化剂,具有更高催化活性及稳定性。
2、本发明的析氧fcs@n-ct复合催化剂制备方法简单、成本低,有利于工业化生产。本发明的析氧fcs@n-ct复合催化剂的制备过程中,n掺杂纳米碳管和fe掺杂cose2纳米颗粒前驱体的合成,以及n掺杂纳米碳管对fe掺杂cose2纳米颗粒前驱体的包覆过程通过一步反应实现,大大简化了工艺步骤,同时实现了n掺杂纳米碳管对fe掺杂cose2纳米颗粒前驱体的原位包覆,使后续的fe掺杂cose2纳米颗粒分散更均匀,稳定性更好。
3、本发明的析氧fcs@n-ct复合催化剂应用于电催化水分解,表现出活性高、稳定性好的特点,相对于商用20wt%pt/c,接近甚至超过商用贵金属ruo2催化剂的催化性能,展现出很好的应用前景。
附图说明
【图1】为实施例1中fcs@n-ct、对比例2中cs、对比例3中c@n-ct、对比例4中fc@n-ct和对比例5中cs@n-ct的xrd图。表明各材料的成功合成。在fcs@n-ct的xrd图中没有发现fe基物质的晶相,表明fe原子只是取代了cose2晶体中的co原子而成功地掺杂在了cose2晶体中。
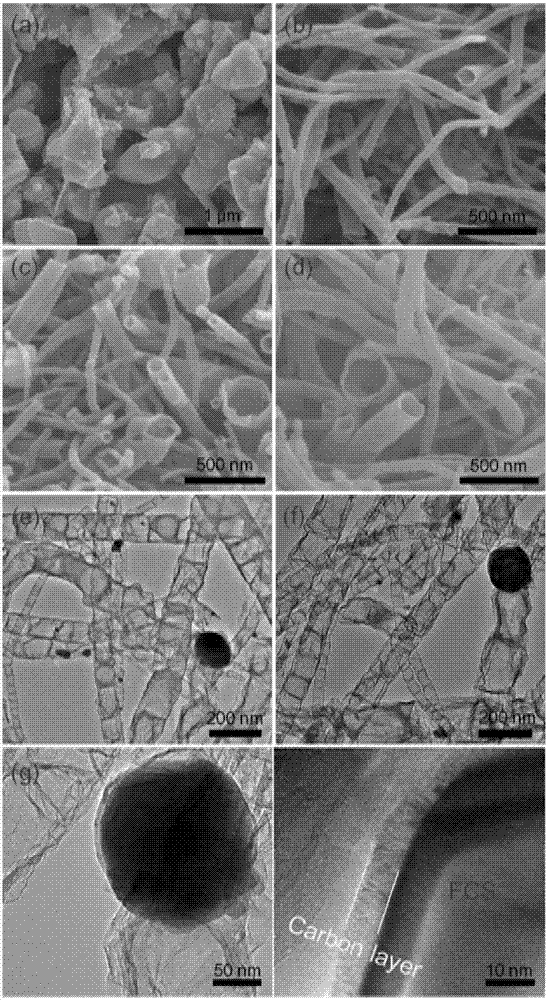
【图2】为对比例2中cs(a)、对比例5中cs@n-ct(b和c)和实施例1中fcs@n-ct(d)的sem图,以及对比例5中cs@n-ct(e)和实施例1中fcs@n-ct(f–h)的tem图。sem图表明cs的微观形貌为完全团聚在一起的颗粒,cs@n-ct和fcs@n-ct呈现出分散的纳米管状结构,最小的管径只有大约20nm,而最大的管径达到280nm左右。tem图进一步显示碳管内部形貌为竹节状,cs和fcs纳米粒子嵌在碳管中且纳米粒子的直径与碳管的直径相当。fcs粒子外存在碳包覆层,碳层的厚度约为6nm。
【图3】为实施例1中fcs@n-ct的(a)xps全谱以及(b)c1s、(c)n1s、(d)co2p、(e)fe2p和(f)se3d的高分辨xps图谱。进一步表明fcs@n-ct的成功合成及fe和n的成功掺杂。
【图4】为实施例1中fcs@n-ct、对比例1中ruo2、对比例2中cs、对比例3中c@n-ct、对比例4中fc@n-ct和对比例5中cs@n-ct在o2饱和的1mkoh溶液中以10mv/s的扫描速率测得的lsv曲线(a),各oer催化剂在10ma/cm处的过电位对比图(b),各oer催化剂的tafel图(c),以及fcs@n-ct和ruo2在1.3–1.7v(vs.rhe)间以100mv/s的扫描速率进行500次cv循环前后测得的lsv对比图(d)。
具体实施方式
下面用实施例更详细地描述本发明内容,但并不限制本发明权利要求的保护范围。
实施例1
co@n-ct(记为c@n-ct)的制备:
采用两步热处理的合成方法,具体过程如下:将0.5gco(ac)2·4h2o与10g尿素在80℃下溶于5ml去离子水中,并在搅拌下蒸发约40min;然后将固体混合物转移到100ml瓷坩埚中,在马弗炉中以5℃/min的加热速率升温至450℃,保温1h,自然冷却;将产品研磨均匀后在高纯n2氛围中以10℃/min的速率加热至800℃,保温1h,自然冷却后得到。
n掺杂的纳米碳管包覆fe掺杂的co纳米粒子(记为fc@n-ct)的制备:
将0.45gco(ac)2·4h2o、0.081gfe(no3)3·9h2o与10g尿素在80℃下溶于5ml去离子水中,其余步骤与上述合成c@n-ct时相同。
cose2@n-ct(记为cs@n-ct)的制备:
取0.5g硒粉和0.1gc@n-ct分别置于加盖瓷舟的上游和下游两端,然后在高纯n2氛围中以20℃/min的速率加热至500℃,保温3h,自然冷却后得到。
fcs@n-ct的制备:
将上述合成cs@n-ct时的0.1gc@n-ct换成0.1gfc@n-ct,其余步骤相同。fcs@n-ct中fe掺杂cose2纳米颗粒的质量百分比含量约为82%,fe掺杂cose2纳米颗粒中fe质量百分比含量约为15%,n掺杂的纳米碳管的质量百分比含量约为18%,n掺杂的纳米碳管中n质量百分比含量约为5%。
采用x射线衍射仪(xrd,rigaku-d/max2500,cu-kα,
样品表层的元素组成采用x射线光电子能谱分析(x-rayphotoelectronspectroscopy,xps),型号为escalab250xi,靶源为al-kα。
工作电极的制备:将5mg样品分散在1ml5wt%nafion溶液/无水乙醇/去离子水(v:v:v=1:3:16)混合液中,超声1h形成均匀的分散液。玻碳电极的预处理步骤如下:首先将一定量的30–50nmal2o3抛光粉置于抛光绒布上,滴加适量的去离子水并打磨约5min,冲洗干净后再在去离子水中超声3–5min;然后在0.2mol/lkno3和1mmol/lk3fe(cn)6的混合溶液中于rst5000电化学工作站上测试循环伏安曲线,扫描电位区间为0–0.6v,扫描速率为50mv/s,所得到的循环伏安曲线中的峰电位差值在80mv以下并尽可能地接近64mv,玻碳电极才能使用,否则重复打磨步骤直到峰电位差值满足要求为止。对于oer电化学性能测试,取10μl分散液滴在直径为3mm的玻碳电极上,自然晾干待用。ruo2电极的制备在相同条件下进行。
所有的电化学测试采用三电极体系,即以pt丝为对电极、饱和甘汞电极(sce)为参比电极和玻碳电极为工作电极。oer测试均在氧气饱和的1mkoh溶液中进行。所有的电位均转换为相对可逆氢电极电位(reversiblehydrogenelectrode,rhe),e(rhe)=e(sce)+0.059×ph+0.242v。
线性扫描伏安法(linearsweepvoltammetry,lsv)通过rst5000电化学工作站进行测试,电位区间为1.2–1.8v(vs.rhe),扫描速率为10mv/s,待测试曲线稳定后进行记录;oer稳定性测试是在扫描电位区间为1.3–1.7v,扫描速率为100mv/s的条件下进行500次cv测试后,重复一次lsv测试并记录。所有的oer测试数据均未经过ir补偿。
fcs@n-ct复合物作为oer催化剂,lsv曲线显示的起始电位为1.53v(vs.rhe),ruo2为1.485v(vs.rhe)(见对比例1)。在电位为1.652v(vs.rhe)时,fcs@n-ct能达到与ruo2(见对比例1)相同的电流密度67.5ma/cm2,而随着电位的继续增加,fcs@n-ct反而比ruo2的电流密度更大。在电流密度为10ma/cm2时,fcs@n-ct所需的过电位η为330mv,ruo2为280mv(见对比例1)。fcs@n-ct的tafel斜率为74mv/dec,低于ruo2(84mv/dec)(见对比例1)。fcs@n-ct在循环500次前后lsv曲线几乎没有变化,而ruo2的lsv曲线在500次cv循环前后达到10ma/cm2的电流密度所需的过电位增加了20mv(见对比例1)。
对比例1
以ruo2为oer催化剂。
ruo2的合成参照wang等人[wangj,yangw,liuj.cop2nanoparticlesonreducedgrapheneoxidesheetsasasuper-efficientbifunctionalelectrocatalystforfullwatersplitting.journalofmaterialschemistrya,2016,4(13):4686–4690]的制备方法。具体步骤如下:将1mmolrucl3·xh2o溶于100ml去离子水中,然后加入1ml1mkoh溶液并在98℃下搅拌1h。自然冷却后用真空抽滤的方法分离固体产物,再用去离子水清洗3次,然后在60℃的真空干燥箱中干燥12h。将前驱体研磨均匀,并在马弗炉中以3℃/min的加热速率升温至300℃煅烧3h,自然冷却后得到的产物即为ruo2。
催化性能的评价方法同实施例1。
ruo2作为oer催化剂,lsv曲线显示的起始电位为1.485v(vs.rhe)。在电位为1.652v(vs.rhe)时,ruo2达到的电流密度为67.5ma/cm2。在电流密度为10ma/cm2时,ruo2所需的过电位η为280mv。tafel斜率为84mv/dec。ruo2在循环500次前后的lsv曲线达到10ma/cm2的电流密度所需的过电位增加了20mv。
对比例2
以cose2(记为cs)为oer催化剂。
cs的制备:
取0.5gco(ac)2·4h2o置于瓷舟中,在高纯n2氛围中以10℃/min的速率加热至800℃,保温1h;然后取0.1g所得到的黑色粉末按实施例1中cs@n-ct的硒化方法取代c@n-ct进行硒化得到cs。
催化性能的评价方法同实施例1。
cs作为oer催化剂,lsv曲线显示的起始电位为1.58v(vs.rhe)。在电位为1.652v(vs.rhe)时,cs达到的电流密度为19.1ma/cm2。在电流密度为10ma/cm2时,cs所需的过电位η为391mv。tafel斜率为106mv/dec。
对比例3
以c@n-ct为oer催化剂。
按实施例1的方法制备c@n-ct。
催化性能的评价方法同实施例1。
c@n-ct作为oer催化剂,lsv曲线显示的起始电位为1.56v(vs.rhe)。在电位为1.652v(vs.rhe)时,c@n-ct达到的电流密度为41.3ma/cm2。在电流密度为10ma/cm2时,cs所需的过电位η为424mv。tafel斜率为111mv/dec。对比例4
以fc@n-ct为oer催化剂。
按实施例1的方法制备fc@n-ct。
催化性能的评价方法同实施例1。
fc@n-ct作为oer催化剂,lsv曲线显示的起始电位为1.59v(vs.rhe)。在电位为1.652v(vs.rhe)时,fc@n-ct达到的电流密度为16.8ma/cm2。在电流密度为10ma/cm2时,fc@n-ct所需的过电位η为400mv。tafel斜率为103mv/dec。
对比例5
以cs@n-ct为oer催化剂。
按实施例1的方法制备cs@n-ct。
催化性能的评价方法同实施例1。
cs@n-ct作为oer催化剂,lsv曲线显示的起始电位为1.61v(vs.rhe)。在电位为1.652v(vs.rhe)时,cs@n-ct达到的电流密度为9.67ma/cm2。在电流密度为10ma/cm2时,cs@n-ct所需的过电位η为350mv。tafel斜率为99mv/dec。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种铝掺杂氧化锌纳米粉体及其微波辅助制备方法与应用
- B掺杂SiC纳米线在场发射阴极材料中的应用
- 一种基于负载型双金属共掺杂纳米复合材料的电化学传感器的制备方法
- 一种楔形Sn掺杂氧化铁纳米棒的制备方法
- 一种汽车电子封装用纳米氧化钇掺杂改性铝镁合金材料及其制备方法
- 具有铁磁性B掺杂g-C<sub>3</sub>N<sub>4</sub>二维超薄纳米片的制备方法
- 基于蛋白生物模板的钆掺杂硫化铜纳米颗粒及其制备方法
- 制备Bi<sub>4</sub>V<sub>2</sub>O<sub>11</sub>纳米晶及银掺杂的BIAGVOX1.0 纳米晶的方法
- 一种可掺杂的钼酸锶纳米材料制备方法
- 一种柔性铝掺杂氧化锌/石墨烯复合材料的制备工艺的制作方法
- 一种氧掺杂碳化氮‑贵金属复合光催化剂、制备方法及应用与流程
- 一种掺铟氧化锌粉体掺杂塑胶专用条状铝颜料的制作方法与工艺
- 一种多孔锌掺杂四氧化三铁/聚苯胺复合材料的制作方法与工艺
- 水热结合锌和氟复合掺杂制备具有优良循环及倍率性能的锰酸锂正极材料的方法与流程
- 一种Ni,Sb‑双掺杂四氧化三钴纳米氧化物的制备方法与流程
- 一种建立仿真模型验证氧化锌掺杂钯对气敏性影响的方法与流程
- 一种Ho3+/Yb3+/Gd3+共掺杂氧化锌上转换发光材料及制备方法与流程
- 一种Ho3+/Yb3+/Li+共掺杂氧化锌上转换发光材料及制备方法与流程
- 一种W与Eu共掺杂的二氧化钒薄膜及其制备方法与流程
- 一种掺铝氧化锌(AZO)中空纳米纤维的制备方法与流程