一种腙键连接型的手性共价有机框架键合硅胶固定相及其应用的制作方法
本发明属于共价有机框架材料领域,具体涉及一种腙键连接型的手性共价有机框架键合硅胶固定相的制备方法及其应用。
背景技术:
共价有机框架(cof)材料是一类由含轻元素(碳、氧、氮、硼等)有机构筑基元通过共价键连接而成的具有周期性网络结构的多孔晶态材料。这类新兴的多孔晶态材料因其具有如质量轻、密度低、结构规整、比表面积大、热稳定性强、骨架大小可调、孔道可修饰等优点使得它们在非均相催化、气体存储、化学传感以及光电材料等领域具有巨大的潜在应用前景。
近年来,cof材料在色谱分离上的应用引起了人们的广泛关注。β-酮胺键连接型cof(如tpbd,chem.commun.,2015,51:12254)、亚胺键连接型cof(如lzu-1,j.chromatogr.a,2016,1436:109和ctppa-1,nat.comm.,2016,7:12104)和硼酯键连接型cof(如cof-1,中国发明专利申请201610058538.7和cof-5,j.chromatogr.a,2016,1445:140)等已被选为新型固定相用于气相色谱和毛细管电色谱分离中,它们对苯同系物,芳香位置异构体以及手性化合物等具有良好的分离效果。然而,新型cof材料作为固定相在高效液相色谱(hplc)分离中研究报道则非常少见。
传统方法制备得到的cof材料颗粒大小不均一,直接将其填充在不锈钢柱管中容易造成柱效低、柱压高和分离能力差等问题。为了解决这一问题,最近,严秀平等成功地制备出了基于cof材料的整体柱,并将其用于hplc分离中,实现了对酚类、苯胺类及苯并噻吩类等化合物的有效分离(j.chromatogr.a,2017,1479:137)。发明人认为解决上述问题的另一策略,就是以大孔硅胶为基质,制备出颗粒大小均匀的cof键合硅胶固定相。然而,到目前为止,将这种cof键合硅胶微球用作hplc的分离中很少报道(j.chromatogr.a,2017,1487:83)。
因此,开发一种颗粒大小均匀的cof键合硅胶固定相具有重大的研究意义和应用价值。
技术实现要素:
本发明的目的在于克服现有技术中的cof材料颗粒大小不均一,直接填充在不锈钢柱管中柱效低、柱压高和分离能力差等缺陷,提供一种腙键连接型的手性共价有机框架键合硅胶固定相。本发明提供的手性共价有机框架键合硅胶固定相,颗粒大小均一,将其应用于高效液相色谱分离中时柱效高、柱压适中、分离能力好。
本发明的另一目的在于提供上述手性共价有机框架键合硅胶固定相在高效液相色谱分离中的应用。
为实现上述发明目的,本发明采用如下技术方案:
一种腙键连接型的手性共价有机框架键合硅胶固定相,所述手性共价有机框架键合硅胶固定相通过如下方法制备得到:在惰性气氛下,先将酰肼手性前体、均苯三甲醛及氨化硅胶在有机溶剂中混合均匀,然后在醋酸催化下发生反应后,过滤、冲洗、干燥,即得所述手性共价有机框架键合硅胶固定相,所述酰肼手性前体的结构如式(ⅰ)
式(ⅰ),
所述氨化硅胶与酰肼手性前体的质量比为2~12:1,酰肼手性前体与均苯三甲醛的摩尔比1:0.5~2。
发明人发现,当选用氨化硅胶为基质键合腙键连接型手性cof材料制备得到的固定相,大小均匀,将其应用于高效液相色谱中时,柱效高、柱压适中,并且通过对几种顺反异构体和位置异构体的分离测试证实,对顺反异构体和位置异构体具有良好的分离效果。
优选地,为了更好地将固定相装填在色谱柱中,优选所述氨化硅胶为球形氨化硅胶。
优选地,所述氨化硅胶的平均孔径优选在100nm或以上。
优选地,所述有机溶剂为二氧六环或二氧六环与均三甲苯的混合溶液。
优选地,所述有机溶剂为体积比为1:0.5~2的二氧六环与均三甲苯的混合溶液。
优选地,所述醋酸与酰肼手性前体的摩尔比为1~20:1;更为优选地,所述醋酸与酰肼手性前体的摩尔比为5~15:1。最为优选地,所述醋酸与酰肼手性前体的摩尔比为7.5:1。
最优选地,所述氨化硅胶与酰肼手性前体的质量比为5.18:1。
酰肼手性前体与均苯三甲醛反应得到的手性cof材料具有二维蜂窝型层状结构,是一种晶型材料。在本发明的比例范围内,都能获得相同结构的手性cof结构。
优选地,所述酰肼手性前体与均苯三甲醛的摩尔比1:1~2。
优选地,所述反应的温度为90~120℃,反应时间为2~5天。
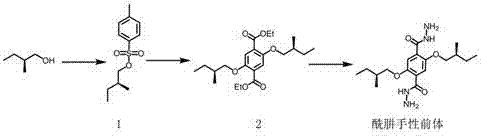
优选地,所述酰肼手性前体的制备方法包括如下制备步骤:
s1:在4-二甲氨基吡啶和三乙胺的作用下,(s)-2-甲基-1-丁醇与对甲苯磺酰氯反应得化合物1:
s2:在碳酸钾的作用下,化合物1与2,5-二羟基对苯二甲酸二乙酯反应,得化合物2:
s3:化合物2和水合肼反应,即得所述酰肼手性前体。
上述手性共价有机框架键合硅胶固定相在高效液相色谱分离顺反异构体或位置异构体中的应用也在本发明的保护范围内。本发明提供的手性共价有机框架键合硅胶固定相应用于高效液相色谱中时,对顺反异构体及位置异构体具有良好的分离效果。
优选地,所述手性共价有机框架键合硅胶固定相在高效液相色谱分离高效氯氰菊酯的顺反异构体、叶菌唑的顺反异构体、硝基甲苯的位置异构体或硝基氯苯的位置异构体中的应用。
与现有技术相比,本发明具有如下有益效果:
本发明提供的腙键连接型的手性共价有机框架键合硅胶固定相,颗粒大小均匀;其应用于高效液相色谱中时柱效高、柱压适中,对顺反异构体和位置异构体具有良好的分离效果,同时本发明的腙键连接型的手性共价有机框架键合硅胶固定相,结构明确,制备方法简单,批次重现性好。
附图说明
图1是本发明实施例1提供的手性共价有机框架键合硅胶固定相的制备流程图;
图2是本发明实施例1提供的酰肼手性前体的制备流程图;
图3是本发明实施例1提供的手性共价有机框架键合硅胶固定相的x射线粉末衍射谱图;
图4是本发明实施例1提供的手性共价有机框架键合硅胶固定相的傅里叶红外光谱图;
图5是本发明实施例1提供的手性共价有机框架键合硅胶固定相的扫描电镜图。
图6是本发明实施例1提供的手性共价有机框架键合硅胶固定相的热重分析曲线。
图7是高效氯氰菊酯(a)、叶菌唑(b)、硝基甲苯(c)和硝基氯苯(d)在本发明实施例1提供的手性共价有机框架键合硅胶固定相上的高效液相色谱图。
图8是高效氯氰菊酯(a)、叶菌唑(b)、硝基甲苯(c)和硝基氯苯(d)在氨化硅胶色谱柱上的高效液相色谱分离图。
具体实施方式
下面结合实施例对本发明做进一步的描述。这些实施例仅是对本发明的典型描述,但本发明不限于此。下述实施例中所用的试验方法如无特殊说明,均为常规方法;所使用的原料,试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。对于所合成的手性共价有机框架键合硅胶固定相的结构,可通过x射线粉末衍射进行表征。在本发明所述的范围内,不同酰肼手性前体与均苯三甲醛比例下所制得的手性共价有机框架键合硅胶固定相均含有相同的手性共价有机框架材料的特征衍射峰,证明本发明所述的范围内,不同酰肼手性前体与均苯三甲醛比例均能获得相同结构的cof键合硅胶固定相,并且表明具有结构明确的二维蜂窝型手性共价有机框架均已成功地键合到了氨化硅胶表面。
实施例1手性共价有机框架键合硅胶固定相
本发明提供的手性共价有机框架键合硅胶固定相由图1所示的流程制备得到。具体地,称取22mg酰肼手性前体、9.8mg均苯三甲醛以及114mg氨化硅胶置于耐压管中,然后加入1.5ml的二氧六环,充分混合均匀后再加入0.15ml醋酸溶液(3mol/l)。将耐压管用氩气置换三次后快速盖上聚四氟乙烯密封塞,然后将其置于预先加热到110℃的烘箱中反应3天。反应结束后,过滤出固体,依次用二氧六环、四氢呋喃和丙酮洗涤,最后置于80℃下真空干燥得到黄色固体粉末130mg,收率为90%。
若在上述制备方法中,不添加氨化硅胶,其余操作步骤与上述一致,可制备得到未键合氨化硅胶的手性共价有机框架材料。
所述酰肼手性前体的制备流程如图2。具体地,包括如下步骤:
(1)化合物1的合成:搅拌下,将对甲苯磺酰氯(3.62g,19mmol)的二氯甲烷50ml缓慢滴加到(s)-2-甲基-1-丁醇(2.0ml,19mmol)、无水三乙胺(5.3ml)和4-二甲氨基吡啶(266mg,1.9mmol)的二氯甲烷溶液中。反应24小时后,往反应混合物中加入100ml水,以二氯甲烷萃取有机相,洗涤、干燥和浓缩后,以柱层析色谱分离(环己烷:二氯甲烷=4:1,v/v)得到化合物1(淡黄色油状液体,3.9g),收率79%。1h-nmr(400mhz,cdcl3)δ(ppm)=7.78(d,j=8.3hz,2h),7.34(d,j=8.5hz,2h),3.87(dd,j=9.3,5.9hz,1h),3.81(dd,j=9.3,6.4hz,1h),2.44(s,3h),1.70(td,j=13.1,6.5hz,1h),1.42–1.34(m,1h),1.17-1.10(m,1h),0.87(d,j=6.8hz,3h),0.82(t,j=7.5hz,3h)。
(2)化合物2的合成:在氩气保护下,化合物1(2.15g,9.0mmol)、2,5-二羟基对苯二甲酸二乙酯(0.915g,3.6mmol)、无水碳酸钾(1.44g,10.8mmol)和n,n-二甲基甲酰胺(50ml)的混合物在90ºc下反应三天。反应结束后,蒸干溶剂,加入水,再用二氯甲烷萃取有机相,浓缩后用柱层析分离(乙酸乙酯:正己烷=25:1),得到化合物2(淡黄色油状液体,980mg),收率70%。1h-nmr(400mhz,cdcl3):δ(ppm)=7.33(s,2h),4.37(q,j=7.2hz,4h),3.86(dd,j=8.7,5.9hz,2h),3.78(dd,j=8.6,6.6hz,2h),1.86(dt,j=13.0,6.5hz,2h),1.62–1.53(m,2h),1.38(t,j=7.1hz,6h),1.31–1.24(m,2h),1.03(d,j=6.7hz,6h),0.94(t,j=7.5hz,6h)。13c-nmr(100mhz,cdcl3):δ(ppm)=166.3,151.6,124.5,116.21,74.3,61.3,34.9,26.0,16.5,14.3,11.3。ms(lc-ms):[m+na]+calcdforc22h34o6na:417.50;found:417.16。
(3)酰肼手性前体的制备:化合物2(860mg,2.2mmol)、水合肼(0.82ml,26.4mmol)和无水乙醇(30ml)的混合物在80ºc下反应24h。反应结束后,冷却到室温,有大量白色固体析出,过滤,干燥即可得到酰肼手性前体(734mg),收率91%。1h-nmr(500mhz,dmso):δ(ppm)=9.16(s,2h),7.38(s,2h),4.56(d,j=3.9hz,4h),3.95(dd,j=9.1,5.8hz,2h),3.86(dd,j=9.1,6.6hz,2h),1.85(dt,j=12.9,6.5hz,2h),1.54–1.47(m,2h),1.29–1.22(m,2h),0.98(d,j=6.7hz,6h),0.91(t,j=7.5hz,6h);13c-nmr(125mhz,dmso):δ(ppm)=163.9,149.9,125.0,114.7,73.9,34.0,25.5,16.3,11.0;ms(lc-ms):[m+h]+calcdforc18h31n4o4:367.47;found:367.16。
所述大孔氨化硅胶(平均孔径为100nm)可参考文献(j.chromatogr.a,2008,1213:162)合成得到,也可以采用市售的其他具有接近孔径的氨化硅胶作为原料。也可以采用市售的其他孔径的氨化硅胶作为原料。
本实施例提供的手性共价有机框架键合硅胶固定相的性能测定结果如下:
(1)x射线粉末衍射测定
图3为本实施例提供的手性共价有机框架键合硅胶固定相的x射线粉末衍射谱图,其中图3a为未键合氨化硅胶的手性共价有机框架材料,图3b为手性共价有机框架键合硅胶固定相,图3c为氨化硅胶。对比三者的x射线粉末衍射谱图,可知本发明成功合成了手性共价有机框架键合硅胶固定相。
(2)傅里叶红外测定
图4为本实施例提供的手性共价有机框架键合硅胶固定相的傅里叶红外光谱图。其中图4a为酰肼手性前体,图4b为均苯三甲醛,图4c为手性共价有机框架材料,图4d为手性共价有机框架键合硅胶固定相。通过对比发现,手性共价有机框架键合硅胶固定相的红外光谱图含有手性共价有机框架材料的特征峰,但不含酰肼手性前体和均苯三甲醛对应的特征峰。
(3)扫描电镜测定
图5本实施例提供的手性共价有机框架键合硅胶固定相的扫描电镜图,其中图5a为手性共价有机框架材料,图5b为手性共价有机框架键合硅胶固定相,图5c为氨化硅胶。对比三者的扫描电镜图,可确定本发明成功地将手性共价有机框架键合到氨化硅胶上制得了手性共价有机框架键合硅胶固定相。
(4)热重分析
图6为本实施例提供的手性共价有机框架键合硅胶固定相在氮气气氛下的热重分析曲线,其中图6a为手性共价有机框架材料,图6b为手性共价有机框架键合硅胶固定相,两者均表现出较好的热稳定性。
(5)高效液相色谱分离
将本实施例提供的手性共价有机框架键合硅胶固定相通过匀浆法填充到150×4.6mmi.d.不锈钢柱管中,应用于顺反异构体和位置异构体的高效液相色谱中。图7是相应的高效液相色谱图,其中a、b分别代表具有顺反异构体的高效氯氰菊酯和叶菌唑,c、d分别代表具有代表位置异构体的硝基甲苯和硝基氯苯。由图可知,本实施例提供的手性共价有机框架键合硅胶固定相可实现所述顺反异构体和位置异构体有效分离。
直接选用氨化硅胶色谱柱(150×4.6mmi.d.)对上述顺反异构体和位置异构体进行高效液相色谱分离时,无法实现有效拆分。如图8所示,即使通过改变流动相,所测试的样品在氨化硅胶柱上均未得到拆分。
- 还没有人留言评论。精彩留言会获得点赞!