催化合成环状碳酸酯的方法及催化剂与流程
本发明涉及一种催化合成环状碳酸酯的方法及其催化剂。
背景技术:
二氧化碳(co2)是一种无色无味无毒的气体。近年来,大气中二氧化碳含量的增加引起全球变暖的问题引起了研究者的广泛关注[1,2]。虽然二氧化碳容易引起温室效应,但它仍具有廉价、易得的优点(参见:x.wang,y.zhou,z.guo,g.chen,j.li,y.shi,y.liu,j.wang,chem.sci.6(2015)6916-6924.,j.langanke,a.wolf,j.hofmann,k.
由co2和环氧化物直接合成环状碳酸酯是一条高效绿色有着百分之百的原子利用率的路线(c.martín,g.fiorani,a.w.kleij,acscatal.5(2015)1353-1370.)。目前,合成环装碳酸酯的非均相催化体系主要为离子液,鎓盐等(参见:x.wang,y.zhou,z.guo,g.chen,j.li,y.shi,y.liu,j.wang,chem.sci.6(2015)6916-6924.,l.-n.he,h.yasuda,t.sakakura,greenchem.5(2003)92-94.,a.sibaouih,p.ryan,m.
技术实现要素:
本发明提供一种可用于催化合成环状碳酸酯的催化剂及这种催化剂的制备方法,同时提供用这种催化剂合成制备环状碳酸酯的方法。
本发明的用于催化合成环状碳酸酯的催化剂为堆积孔结构的fe4ⅲ(fe(cn)6)3,或者为堆积孔结构的km4(feⅲ(cn)6)3,其中m为co或ni或zn。
本发明所述的催化剂fe4ⅲ(fe(cn)6)3制备方法是:将fecl2·4h2o和k3[fe(cn)6]·6h2o分别溶蒸馏水中,将以上两种溶液在搅拌下混合,再将混合物在80℃加热12小时,通过离心分离得到的固体,用高纯水充分洗涤所得到固体后在80℃下干燥处理得到所述的催化剂。
本发明所述的催化剂km4(feⅲ(cn)6)3制备方法是:将fecl2·4h2o(co(no3)2·6h2o,ni(no3)2·6h2o,zncl2)和k3[fe(cn)6]·6h2o分别溶蒸馏水中,将以上两种溶液在搅拌下混合,再将混合物在80℃加热12小时,通过离心分离得到的固体,用高纯水充分洗涤所得到固体后在80℃下干燥处理得到所述的催化剂。
本发明的催化合成环状碳酸酯的方法是:在反应容器中加入环氧化物的反应底物、用所述的催化剂fe4ⅲ(fe(cn)6)3或km4(feⅲ(cn)6)3和共催化剂,常压下并将反应体系在60~100℃油浴中搅拌反应,反应完成后通过过滤收集产物—环状碳酸酯,所用的共催化剂为任一种季铵盐。
优选地,本发明的催化合成环状碳酸酯的方法是所用的共催化剂为tbab。
优选地,本发明的催化合成环状碳酸酯的方法是所用的共催化剂为tbac。
优选地,本发明的催化合成环状碳酸酯的方法是所用的共催化剂为tbai。
本发明所述的催化合成环状碳酸酯的方法中,所用的底物可以是氧化苯乙烯、环氧氯丙烷、甲基丙缩水甘油酯、正丁基缩水甘油醚、氧化环己烯或苯基缩水甘油醚的任一种。
相关的试验表明,本发明的催化剂在常压、无溶剂的条件下对催化二氧化碳和环氧化物的环状反应表现较高的催化活性,可以重复循环使用,且其制备方法非常简单,一种高效,绿色的铁络合物催化剂。采用本发明的方法可以在温和条件下(无溶剂和100℃)高转化率和选择性的得到产物。此外,本发明的催化剂在反应结束后可以很好的和底物进行分离,方便了催化剂的重复利用;本发明的反应过程中不需要溶剂,免去了反应结束后溶剂的分离步骤,而且反应过程中二氧化碳压力为常压,因此也不再需要附加的加压设备。
附图说明
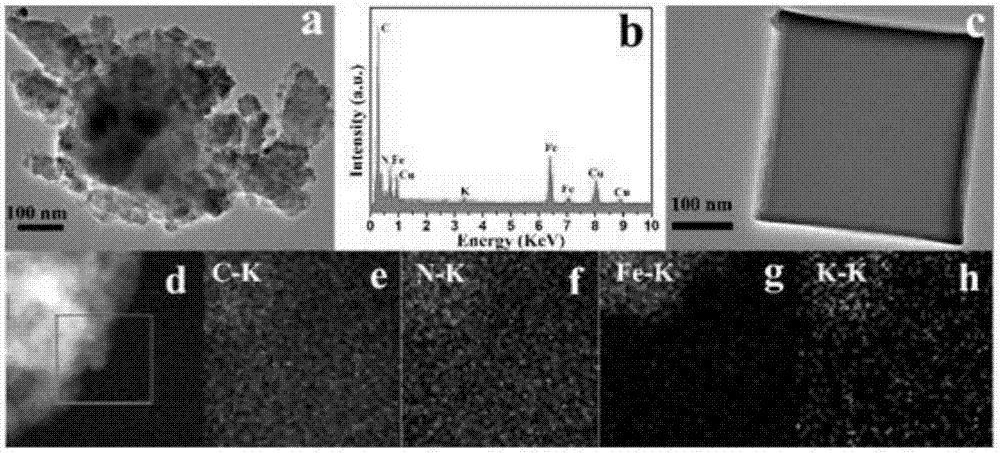
附图1为本发明fe4ⅲ(fe(cn)6)3催化剂的电镜、能谱图。其中:(a)为透射电镜图,(b)为其能量色散x射线光图谱,(c)是对比催化剂feⅲ4(fe(cn)6)3-cube的透射电镜图,(d)大角度环形暗场扫描透射电子显微镜,(e-h)为feⅲ4(fe(cn)6)3元素分布图。
附图2为feⅲ4(fe(cn)6)3氮气吸脱及孔体积曲线,其中:(a)feⅲ4(fe(cn)6)3和feⅲ4(fe(cn)6)3-cube氮气吸脱附曲线,(b)feⅲ4(fe(cn)6)3和feⅲ4(fe(cn)6)3-cube的孔体积。
附图3为feⅲ4(fe(cn)6)3x衍射谱图及精细衍射谱图,其中:(a)所有元素feⅲ4(fe(cn)6)3的xps全谱,(b)fe元素的xps精细谱。
附图4为feⅲ4(fe(cn)6)3x衍射花样及拉曼谱图,其中:(a)为x衍射花样,(b)为拉曼光谱图。
附图5为feⅲ4(fe(cn)6)3循环利用后转化率与选择性的关系曲线。
具体实施方式
以下结合实施例对本发明进行解说。
一、催化剂的制备
1、催化剂的制备
1.1feⅲ4(fe(cn)6)3的制备
将fecl2·4h2o(1mmol)和k3[fe(cn)6]·6h2o(0.5mmol)分别溶于20ml蒸馏水中。将以上两种溶液在磁力搅拌下混合,将混合物转移到聚四氟乙烯内衬的不锈钢高压釜中并在80℃加热12小时。最后,通过离心分离得到的固体用高纯水洗涤3次并在80℃下干燥12小时,得到feⅲ4(fe(cn)6)3。
1.2km4(feⅲ(cn)6)3(fe(cn)6)3-cube的制备
将co(no3)2·6h2o,·6h2o(1mmol)和k3[fe(cn)6]·6h2o(0.5mmol)分别溶于20ml蒸馏水中。将以上两种溶液在磁力搅拌下混合,将混合物转移到聚四氟乙烯内衬的不锈钢高压釜中并在80℃加热12小时。最后,通过离心分离得到的固体用高纯水洗涤3次并在80℃下干燥12小时,得到kco4(feⅲ(cn)6)3。
将zncl2·6h2o(1mmol)和k3[fe(cn)6]·6h2o(0.5mmol)分别溶于20ml蒸馏水中。将以上两种溶液在磁力搅拌下混合,将混合物转移到聚四氟乙烯内衬的不锈钢高压釜中并在80℃加热12小时。最后,通过离心分离得到的固体用高纯水洗涤3次并在80℃下干燥12小时,得到kzn4(feⅲ(cn)6)3。
将ni(no3)2·6h2o(1mmol)和k3[fe(cn)6]·6h2o(0.5mmol)分别溶于20ml蒸馏水中。将以上两种溶液在磁力搅拌下混合,将混合物转移到聚四氟乙烯内衬的不锈钢高压釜中并在80℃加热12小时。最后,通过离心分离得到的固体用高纯水洗涤3次并在80℃下干燥12小时,得到(kni4(feⅲ(cn)6。
1.3fe(cn)6)3-cube的制备
本发明后述的(fe(cn)6)3-cube的制备可参见l.zhang,h.b.wu,x.w.lou,j.am.chem.soc.135(2013)10664-10672.,即]在恒定的磁力搅拌下将0.5mmolk4fe(cn)6·3h2o加入到hcl溶液(0.1m,50ml)中搅拌10分钟后,得到澄清溶液。然后将混合物转移到聚四氟乙烯内衬的不锈钢高压釜中并在80℃下加热12小时。最后通过离心收集所得的固体并用超纯水洗涤3次,然后在80℃下干燥12h。
2、催化剂表征
对前所制备的催化剂的形貌和微观结构进行透射电镜(tem)表征,参见附图1。
feⅲ4(fe(cn)6)3的tem图像(图1a)表明,该催化剂为团簇形貌。为了研究feⅲ4(fe(cn)6)3的元素分布,对催化剂进行了大角度环形暗场扫描透射电子显微镜(haadf-stem),参见图1d,催化剂所含有的元素分别为碳、氮、铁。催化剂中元素分布图1b分析表明催化剂中c,n,k和fe均匀分布,此外还提供了催化剂中元素的摩尔分数(c83.55%,n10.50%,k0.33%和fe3.39%)。图1c显示了feⅲ4(fe(cn)6)3-cube为立方结构。mapping图谱的图1e-图1h表明了催化剂的元素分布较均匀。
为了表征催化剂的比表面积,本发明对催化剂进行bet测试,参见附图2。
由图2a可以看出吸脱附曲线符合iv型吸附等温线,表明该催化剂为堆积孔结构[24]。feⅲ4(fecn)6)3的比表面积和孔体积分别为296.3m2/g和0.23cm3/g。大表面积和孔体积可以为催化剂提供更多暴露的活性位点[25,26]。feⅲ4(fe(cn)6)3-cube的吸脱附曲线表明该催化剂不具有介孔结构。feⅲ4(fe(cn)6)3-cube的表面积和孔体积分别为10.0m2/g和0.018cm3/g。小的比表面积以及孔体积表明feⅲ4(fe(cn)6)3-cube催化剂为实心立方体结构,该结构特性不利于活性位点与底物的接触。
为了表征催化剂的元素的价态分布,对催化剂进行xps测试,参见附图3和图4。
从xps全谱(图3a)清晰地表明在催化剂表面上有c1s(284.6ev),n1s(397.9ev)和fe2p(708.5和721.8ev)元素。fe2p的精细谱图(图3b)由四个主峰组成,其结合能分别为708.5,711.5,721.4和724.3ev。708.5和724.3ev的峰对应于[feii(cn)6]的fe2p3/2和fe2p1/2,711.5和721.4ev的结合能对应于feⅲ4(fe(cn)6)3中的feⅲ2p3/2和feⅲ2p1/2。
从图4a可知,晶格面的催化剂的xrd图谱显示在17.3°,24.6°,35.2°,39.4°,43.4°,50.5°,53.9°和57.0°8个特征峰为催化剂的(200),(220),(400),(420),(422),(440),(600)和(620)晶面。图4b所示为拉曼光谱,在2091和2130cm-1两个强峰为碳氮三键的伸缩振动。此外,在264和534cm-1处的谱带为fe-c≡n-fe的伸缩和弯曲振动峰。
二、催化剂的评估
化剂催化性能的测试在常压下25ml三口圆底烧瓶中进行。在三口圆底烧瓶中加入环氧化物(5mmol),催化剂(10mg)和共催化剂(bu4nbr或其它)2.5mol%,并将反应体系在油浴中以600rpm/min的转速进行搅拌,反应时间为0.25-5小时。反应后通过过滤收集产物,并通过气相色谱质谱仪(gc-ms,agilent6,890n/5,937n)和1hnmr(使用cdcl3作为溶剂的varianinova400,400mhz)来检测转化率,选择性和产物收率。
催化剂的活性计算为:转化率%=100×([c0-c1]/[c0]),选择性%=100×[c”]/[c’]和产率%=100×[c”]/[c0],[c0]为初始底物浓度,[c1]为反应结束时底物浓度,[c’]产物浓度[c”]是目标产物的浓度。
以氧化苯乙烯与co2环加成生成苯乙烯碳酸酯作为模型反应对催化剂进行评估,催化结果如表1所示。
当反应在不使用共催化剂的条件下进行时(表1,第1项)或在100℃和1atmco2压力下仅使用tbab(表1,第2项)时,只有少量的产物。该过程表明催化剂和共催化剂对于环加成反应是必不可少的。第3-9项显示不同类型的催化剂具有不同的催化活性。feⅲ4(fe(cn)6)3催化剂在共催化剂(tbab)存在下有着转化率和选择性都是99%的良好催化效果。用tbac(第8项)或tbai(第9项)作为共催化剂时feⅲ4(fe(cn)6)3的催化效果不能达到前者的良好效果。由以上研究可以得出该反应中路易斯酸和亲核试剂是该反应的必须条件。三种共催化剂的区别在于卤素离子的不同,因此共催化剂对环加成反应主要是卤素离子的影响。由表1可知活性按tbab>tbai>tbac的顺序递减,这归因于卤化物阴离子的亲核性质(cl->br->i-)和离去能力(i->br->cl-)。从3,10-13中可以发现温度对环加成反应的影响也较大。随着温度的降低,转化率和选择性也会随之降低。较低的反应温度会产生较多的副产物,因此,较高的温度有利于实现更好的产率。
表1氧化苯乙烯和co2与不同催化剂反应的结果
反应条件:苯乙烯氧化物(5mmol),2.5mol%共催化剂,催化剂(10mg),co2压力(1atm),反应时间(3h),gc-ms测定转化率和选择性。
为了研究催化剂的广谱性,本发明对其他类型的环氧化物在1atm压力和100℃下进行环加成反应。不同类型的环氧化物转化为相关的产物及转化率、选择性参见表2。由表2可知大部分底物都有着良好的转化率和选择性。在tbab和催化剂存在下,环氧氯丙烷的转化率和选择性都为99%,主要是氯原子的吸电子能力该反应的进行。环己烯氧化物的转化效果不好,这可能是由于空间位阻造成的。
表2环氧化物和co2的环加成反应
a反应条件:通过gc-ms测定环氧化物(5mmol),2.5mol%催化剂,催化剂(10mg),co2压力(1atm),反应时间(3h),转化率和选择性。b反应时间(5小时)
三、催化剂回收再利用
三、催化剂回收再利用
一个良好催化剂不仅要有良好的催化活性,还应具有良好的可回收性重复利用性。本发明对催化剂的循环性在一个大气压的二氧化碳和氧化苯乙烯在tbab作共催化剂100℃无溶剂的条件进行,反应完成后对催化剂进行离心收集并用乙醇洗涤三次,然后真空干燥催化剂以便重新用于下一次反应。催化剂的重复性能如图5所示,可以看出经过五次循环后,催化剂的催化性能有略微的降低。这个结果表明该催化剂有良好的可回收性重复利用性。最后,我们用icp对循环五次后的催化剂进行检测,发现fe的含量从37.39%下降到33.92%。
- 还没有人留言评论。精彩留言会获得点赞!