一种膦氮配位型金属催化剂及其应用的制作方法
本发明属于金属有机催化剂领域,具体涉及一种膦氮配位型金属催化剂及其应用。
背景技术:
α-烯烃是一种非常重要的有机原料和化学中间体,被广泛用于合成共聚聚乙烯、表面活性剂、高级合成润滑油(聚α-烯烃)、增塑剂和精细化学品。
其中,1-己烯和1-辛烯主要用作聚乙烯的共聚单体,1-辛烯也被用作聚α-烯烃(pao)的原料。随着高端聚烯烃(poe)和合成润滑油(聚α-烯烃)需求量的不断增长,1-己烯与1-辛烯则成为α-烯烃市场的主要增长点,占总需求的30%左右。
相比于传统的蜡裂解、萃取分离、烷烃催化裂解、烷烃脱氢、煤品抽提、脂肪醇脱氢、内烯烃异构等方法,乙烯选择性齐聚法则为生产1-己烯与1-辛烯提供了一条清洁高效的路径。
美国专利us3300458最早报道了乙烯选择性三聚的现象,欧洲专利ep0417477报道了由2,5-二甲基吡咯、2-乙基己酸铬、三乙基铝和二乙基氯化铝组成的乙烯三聚催化体系,并在卡塔尔成功实现了1-己烯的工业化生产。
国内工业利用乙烯齐聚生产1-己烯的装置较少,目前,仅有10万吨/年的产能。现有乙烯齐聚工艺中包括催化剂制备、齐聚、产品分离三个部分。
现有乙烯齐聚工艺中,催化剂的制备以间歇方式进行,催化剂各组分在高纯氮保护下与来自脱水塔的脱水环己烷在催化剂配制釜中稀释到一定浓度,然后通过各自的计量泵控制流量,按照给定的配比关系注入到反应系统中;反应温度115℃,反应压力5.0mpa,停留时间1.0h,产品分布:丁烯0.07wt%、1-己烯94.39wt%、辛烯0.29wt%、癸烯5.25wt%,该催化剂体系可以高选择性生产1-己烯,却无法选择性生产1-辛烯。
另有carter等(highactivityethylenetrimerisationcatalystsbasedondiphosphineligands.chem.commun.2002,858)报道了一种铬催化剂,利用三氯化铬、pnp配体(p上为甲氧基苯基)和甲基铝氧烷(mao)组成的体系,1-己烯的选择性接近90%,催化活性为1030kg/(gcr·h),但是,该催化剂体系无法高选择性生产1-辛烯,且其配体的合成步骤较为繁琐。
利用乙烯齐聚高选择性制备1-辛烯的报道相对较少,bollmann等(ethylenetetramerization,anewroutetoproduce1-octeneinexceptionallyhighselectivities.j.am.chem.soc.2004,126,14712)报道,在乙烯齐聚反应中,由(r2)2pn(r1)p(r2)2(pnp)配体、三氯化铬和甲基铝氧烷(mao)组成的催化剂,1-辛烯的选择性最高为70wt%左右,端烯烃的选择性在90%以上,然而,该乙烯四聚体系的选择性并没有达到乙烯三聚超过90%的选择性。
现有技术中通过改变pnp配体的骨架结构,得到了一系列衍生的基于p,p配位的配体,如pnnp骨架配体、pcnp骨架配体、pncnnp骨架配体、pcncp骨架配体等,与铬源组成催化体系,但是,其催化反应活性和1-辛烯的选择性并没有得到明显提升,而配体的合成步骤却更为复杂。
目前,国内尚没有实现乙烯四聚制备1-辛烯的工业生产,也没有特别成熟的催化剂体系。
技术实现要素:
本发明的目的在于提供一种膦氮配位型金属催化剂及其应用,该催化剂的配体具有两种配位原子,催化活性高,稳定性良好,利用该膦氮配位型金属催化剂,可以实现高选择性联产1-己烯和1-辛烯,两种线性α-烯烃的质量分数之和最高可超过95%;通过对膦氮基团的调变,还能够实现1-己烯和1-辛烯两种产品不同产出比的高选择性合成。
为了实现上述目的,本发明提供如下技术方案:
一种膦氮配位型金属催化剂,其以pcmn为主体骨架,化学通式为[r1r2pcmnh(r3)]mxn,m为2或3,结构式如式i所示:
其中,cm为烷基、取代烷基、芳基、取代芳基或其他含不饱和键的碳桥联基团,m代表桥联碳原子的个数,m为2或3;r1、r2为给电子基团或吸电子基团,种类相同或不同,r1、r2分别独立地代表直链烷基、支链烷基、杂烷基、环烷基、杂环烷基、芳基、取代芳基或含不饱和键的基团;r3为氢、烷基、取代的烷基、环烷基、杂环烷基、芳基、取代芳基或其他含不饱和键的基团;mxn为金属化合物,金属m为ti、zr、hf、v、cr、fe、co或ni中的一种;x为f、cl、br、i或羰基,n为2-6。
优选地,所述cm选自–ch2–ch2–、–ch(ch3)–ch2–、–ch(ch3)–ch(ch3)–、–ch(ph)–ch2–、–ch(ph)–ch(ph)–、–c6h4–、–c6h4–ch2–、–ch2–ch2–ch2–、–ch(ch3)–ch2–ch2–、–ch(ph)–ch2–ch2–和–ch(ph)–ch(ph)–ch2–中的一种。
优选地,r1、r2分别选自正丁基、异丁基、叔丁基、苯基、2-甲基苯基、3-甲基苯基、4-甲基苯基、2,6-二甲基苯基、2,6-二乙基苯基、2,6-二异丙基苯基、2,4,6-三甲基苯基、2-甲氧基苯基、4-甲氧基苯基、2,6-二甲氧基苯基、2,6-二乙氧基苯基、2,4,6-三甲氧基苯基、萘基、联苯基、吡咯基、哌啶基、2-噻吩基、2-呋喃基、2-吡啶基和3-吡啶基中的一种。
优选地,r3选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、环丁基、环戊基、环己基、2-甲基环己基、2,6-二甲基环己基、金刚烷基、乙烯基、烯丙基、苯基、苄基、萘基、2-甲基苯基、3,5-二甲基苯基、3,5二甲氧基苯基、3,5-二异丙基苯基、2-噻吩基、2-呋喃基、2-吡啶基和4-吡啶基中的一种。
又,金属化合物mxn选自三氯化铬、三氯化铬的四氢呋喃复合物、二氯化铬、二氯化铬的四氢呋喃复合物、二氯化铬的甲苯四氢呋喃复合物、二氯化铬的碳卡宾复合物、三氯化铬的碳卡宾复合物、乙酰丙酮铬、三(2-乙基己酸)铬、甲基二氯化铬四氢呋喃复合物、三苯基铬四氢呋喃复合物、二甲基铬碳卡宾复合物、二乙基铬碳卡宾复合物、二苯基铬碳卡宾复合物、羰基铬、氯化镍和烷基镍中的一种。
本发明提供所述膦氮配位型金属催化剂在乙烯齐聚联产1-己烯和1-辛烯中应用。
一种通过乙烯齐聚反应制备1-己烯和1-辛烯的方法,其包括:在乙烯氛围下,将权利要求1所述膦氮配位型金属催化剂、活化剂加入有机溶剂,将乙烯升压,进行齐聚反应;反应温度为0~180℃,反应压力0.1~18mpa,反应时间0.01~48h;其中,膦氮配位型金属催化剂的摩尔浓度为0.001~100mmol/l,活化剂的摩尔浓度为0.1~1000mmol/l,所述活化剂为烷基铝化合物或有机硼化合物。
优选地,所述有机溶剂选自甲苯、二甲苯、均三甲苯、戊烷、环戊烷、甲基环戊烷、己烷、环己烷、甲基环己烷、庚烷、辛烷、壬烷、癸烷、碳十一烷、碳十二烷、碳十三烷、碳十四烷、碳十五烷、碳十六烷、1-己烯、1-辛烯、1-癸烯、二氯甲烷、二氯乙烷、氯苯、邻二氯苯、间二氯苯、对二氯苯、溴苯、邻二溴苯、间二溴苯、对二溴苯和碘苯中的一种。
又,所述烷基铝化合物选自三甲基铝、三乙基铝、三异丁基铝、三正丁基铝、三正己基铝、三正辛基铝、甲基铝氧烷、乙基铝氧烷、异丁基铝氧烷及其改性铝氧烷、二乙基氯化铝、乙基二氯化铝、三(五氟苯基)铝中的一种或几种的混合物。
优选地,所述有机硼化合物选自三(五氟苯基)硼、四氟硼酸盐、四(五氟苯基)硼酸盐、三全氟芳基硼烷和四全氟芳基硼酸盐中的一种或几种的混合物。
本发明根据乙烯选择性齐聚的金属环化机理,开发新型结构的催化剂,实现1-己烯和1-辛烯两种产品的高选择性联产,作为乙烯选择性齐聚领域的优选路线。
本发明的膦氮配位型金属催化剂,以pcmn为主体骨架,配体具有p和n两种配位原子,取代基团r1、r2和r3可以调变,可以是给电子基团也可以是吸电子基团,r1和r2的种类可以相同,也可以不同,取代基的调变在很大范围内调控金属中心周围的电子效应和空间位阻效应,从而不同程度地稳定金属中心;改变p和n上的取代基,催化剂的催化性能也随之发生改变,这可能是催化剂活化过程中形成了不同的具有配位空位的活性物种。由于金属中心周围的电子效应和空间位阻效应的不同,乙烯分子与活性物种金属中心配位插入的能力也不同,从而造成催化性能的差异。
与现有技术相比,本发明具有如下有益效果:
本发明的膦氮配位型金属催化剂,以pcmn为主体骨架,具有p和n两种配位原子,催化活性高,其催化活性在0.1×106~90×106g/(molcr·h)范围内;且催化剂稳定性良好,持续反应180分钟后,其催化活性仍维持在0.1×106~90×106g/(molcr·h),催化产物中1-己烯和1-辛烯的质量分数之和基本保持不变。
利用本发明的膦氮配位型金属催化剂,进行乙烯齐聚反应时,可以高选择性地联产1-己烯和1-辛烯,随着膦氮原子上取代基团的不同,或者反应温度的不同,还可以得到不同比例的1-c6和1-c8,1-c8/1-c6在1~4:1范围内变化,两种线性α-烯烃的质量分数之和在90%以上。
附图说明
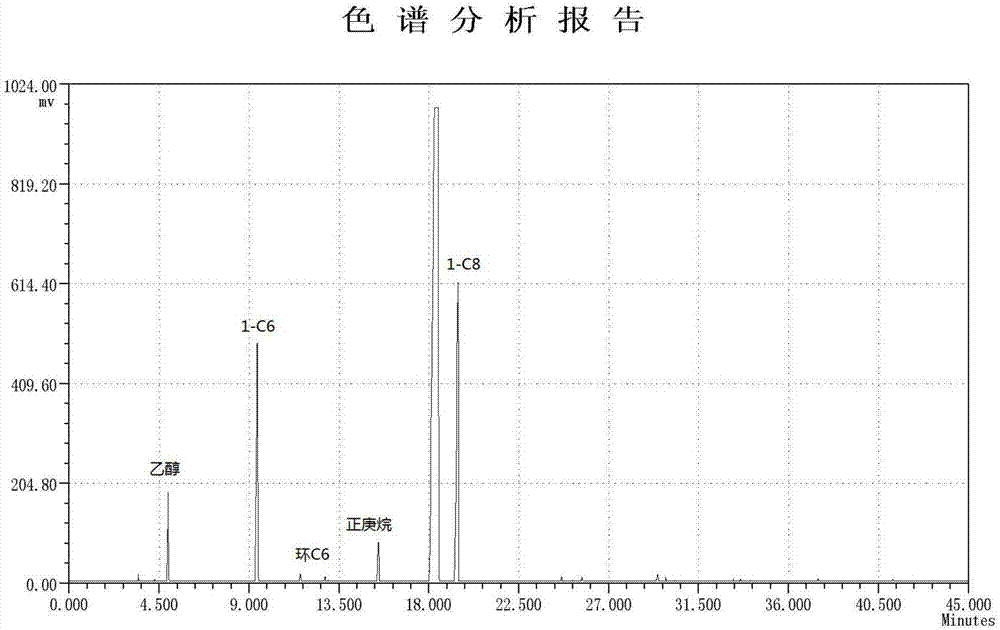
图1为本发明实施例2齐聚产物的气相色谱分析图(内标物为正庚烷)。
图2为本发明实施例18中制备的膦氮配位型铬催化剂的分子结构图。
图3为本发明实施例30齐聚产物的气相色谱分析图(内标物为正庚烷)。
具体实施方式
以下结合具体实施里对本发明做进一步说明。
实施例1一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式ii所示膦氮配位型金属催化剂的甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml、甲基铝氧烷甲苯溶液1ml(1.5mol/l)、式ii所示膦氮配位型金属催化剂甲苯溶液20ml,快速加热至80℃,同时提升乙烯压力至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入150ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标物,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式ii所示膦氮配位型金属催化剂活性为6.8×106g/(molcr·h),产物组成记录于表1。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例2
将实施例1中的反应温度由80℃调变至70℃,其他操作条件不变,利用内标法计算得到催化剂活性为12.6×106g/(molcr·h),产物组成记录于表1,产物的气相色谱分析见附图1。
实施例3
将实施例1中的反应温度由80℃调变至60℃,其他操作条件不变,利用内标法计算得到催化剂活性为16.5×106g/(molcr·h),产物组成记录于表1。
实施例4
将实施例1中的反应温度80℃调变至50℃,其他操作条件不变,利用内标法计算得到催化剂活性为12.3×106g/(molcr·h),产物组成记录于表1。
实施例5
将实施例1中的反应温度80℃调变至40℃,其他操作条件不变,利用内标法计算得到催化剂活性为8.1×106g/(molcr·h),产物组成记录于表1。
实施例6
将实施例3中的乙烯压力由4mpa变至3mpa,其他操作条件不变,利用内标法计算得到催化剂活性为11.8×106g/(molcr·h),产物组成记录于表1。
实施例7
将实施例3中的乙烯压力由4mpa变至2mpa,其他操作条件不变,利用内标法计算得到催化剂活性为6.6×106g/(molcr·h),产物组成记录于表1。
实施例8
将实施例3中的乙烯压力4mpa变至1mpa,其他操作条件不变,利用内标法计算得到催化剂活性为2.8×106g/(molcr·h),产物组成记录于表1。
实施例9
将实施例3中的反应时间由60min变至120min,其他操作条件不变,利用内标法计算得到催化剂活性为13.6×106g/(molcr·h),产物组成记录于表1。
实施例10
将实施例3中的反应时间由60min变至180min,其他操作条件不变,利用内标法计算得到催化剂活性为12.1×106g/(molcr·h),产物组成记录于表1。
表1实施例1~10中的催化反应结果
由表1可见,随着反应温度由80℃降至40℃,催化反应活性先升高后降低,1-己烯的选择性逐渐降低而1-辛烯的选择性逐渐升高;随乙烯压力由4mpa降至1mpa,催化反应活性呈下降趋势,1-己烯的选择性逐渐升高而1-辛烯的选择性逐渐降低;随着反应时间由60分钟延长至180分钟,催化反应活性略有下降,1-己烯和1-辛烯的选择性基本不变。因此,催化反应条件,包括反应温度和乙烯压力,对催化活性和产物选择性均有明显的调控作用。
实施例11一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式iii所示膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式iii所示膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式iii所示膦氮配位型金属催化剂活性为8.7×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例12一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式iv所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式iv所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式iv所示膦氮配位型金属催化剂活性为3.2×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例13一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式v所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式v所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式v所示膦氮配位型金属催化剂活性为6.5×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例14一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式vi所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式vi所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式vi所示的膦氮配位型金属催化剂活性为5.3×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例15一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式vii所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式vii所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式vii所示的膦氮配位型金属催化剂活性为9.5×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例16一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式viii所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式viii所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式viii所示的膦氮配位型金属催化剂活性为3.8×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例17一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式ix所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式ix所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式ix所示的膦氮配位型金属催化剂活性为6.8×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例18一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式x所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式x所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到催化剂活性为19.6×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
所述的膦氮配位型铬催化剂的制备过程为:将前体2-ph2pc6h4ch2nh2和前体crcl3(thf)3溶于四氢呋喃溶液中,室温搅拌12h,减压除去溶剂,固体粗产物经正己烷洗涤,真空干燥,得到所述催化剂。
制备的铬催化剂分子结构中,cr分别与p、n、三个cl和一个四氢呋喃配位,形成六配位的八面体配位几何,晶体结构参见附图2,配位的溶剂分子在活化剂的作用下离去,形成具有配位空位的活性物种。
实施例19一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式xi所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式xi所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式xi所示的膦氮配位型金属催化剂活性为12.3×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例20一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式xii所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式xii所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式xii所示的膦氮配位型金属催化剂活性为11.2×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例21一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式xiii所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式xiii所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式xiii所示的膦氮配位型金属催化剂活性为9.6×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
实施例22一种通过乙烯齐聚反应生产1-己烯和1-辛烯的方法
在手套箱中新鲜配制式xiv所示的膦氮配位型金属催化剂甲苯溶液20ml,金属催化剂的摩尔浓度为0.25mmol/l,备用。
将高压反应釜安装调试完毕,预热至100℃,真空干燥5h后冷却至室温,置换为乙烯氛围,依次注入甲苯溶液60ml,甲基铝氧烷甲苯溶液1ml(1.5mol/l),式xiv所示的膦氮配位型金属催化剂甲苯溶液20ml,快速加热至60℃,同时将乙烯压力升至4mpa,搅拌速率500rpm,保持60min。
快速冷却反应体系至0℃,泄压,加入100ml无水乙醇和5ml10%盐酸的混合液进行淬灭,震荡,静置,加入2g正庚烷内标,取干燥后的有机相液体进行气相色谱分析。
利用内标法计算得到式xiv所示的膦氮配位型金属催化剂活性为8.5×106g/(molcr·h),产物组成记录于表2。
其中,所述的膦氮配位型金属催化剂结构式如下:
表2实施例11~22中的催化反应结果
由表2可见,随着p或n上取代基的调变,催化反应活性和产物选择性均发生明显的变化,取代基的电子效应和空间位阻效应均可影响金属中心的化学环境,从而调变催化剂的性能。
实施例23
采用实施例1催化剂,配位金属换成crcl2,其他条件不变,用于乙烯齐聚反应。利用内标法计算得到催化剂活性为1.5×106g/(molcr·h),产物组成记录于表3,催化剂结构如下:
实施例24
采用实施例16催化剂,配位金属换成crph3,其他条件不变,用于乙烯齐聚反应。利用内标法计算得到催化剂活性为1.2×106g/(molcr·h),产物组成记录于表3,催化剂结构如下:
实施例25
采用实施例19催化剂,配位金属换成crme2,其他条件不变,用于乙烯齐聚反应。利用内标法计算得到催化剂活性为0.6×106g/(molcr·h),产物组成记录于表3,催化剂结构如下:
实施例26
采用实施例21催化剂,配位金属换成cr(co)6,在两倍当量ag[al(oc(cf3)3)4]和500当量三乙基铝(tea)作用下进行乙烯齐聚反应。利用内标法计算得到催化剂活性为0.2×106g/(molcr·h),产物组成记录于表3,催化剂结构如下:
实施例27
采用实施例11催化剂,配位金属换成nicl2,其他条件不变,用于乙烯齐聚反应。利用内标法计算得到催化剂活性为0.3×106g/(molni·h),产物组成见表3,催化剂结构如下:
实施例28
采用实施例20催化剂,配位金属换成nicl2,其他条件不变用于乙烯齐聚反应。利用内标法计算得到催化剂活性为0.4×106g/(molni·h),产物组成见表3,催化剂结构如下:
表3实施例23~28的催化反应结果
由表3可见,调变催化剂的金属中心,包括元素类型或金属的配位基团,催化反应活性和产物选择性均发生明显变化,说明金属中心本身的性质对催化剂性能具有至关重要的影响。
实施例29
将实施例18中的甲基铝氧烷甲苯溶液加入量由1ml改为3ml,其他操作条件不变,利用内标法计算得到催化剂活性为22.8×106g/(molcr·h),产物组成记录于表4。
实施例30
将实施例18中的甲基铝氧烷甲苯溶液加入量由1ml改为3ml,反应温度由60℃改为30℃,其他操作条件不变,利用内标法计算得到催化剂活性为2.4×106g/(molcr·h),产物组成记录于表4,产物的色谱分析图谱参见附图3。
实施例31
将实施例18中的甲基铝氧烷甲苯溶液改为三乙基铝/四(五氟苯基)硼酸盐甲苯溶液,其他操作条件不变,利用内标法计算得到催化剂活性为1.5×106g/(molcr·h),产物组成记录于表4。
实施例32
将实施例18中的甲基铝氧烷甲苯溶液改为三正丁基铝/四(五氟苯基)硼酸盐甲苯溶液,其他操作条件不变,利用内标法计算得到催化剂活性为2.3×106g/(molcr·h),产物组成记录于表4。
实施例33
将实施例18中的甲基铝氧烷甲苯溶液改为三异丁基铝/四(五氟苯基)硼酸盐甲苯溶液,其他操作条件不变,利用内标法计算得到催化剂活性为2.8×106g/(molcr·h),产物组成记录于表4。
实施例34
将实施例18中的甲苯溶液换成环己烷溶液,其他操作条件不变,利用内标法计算得到催化剂活性为15.3×106g/(molcr·h),产物组成记录于表4。
实施例35
将实施例18中的反应压力由4mpa变为5mpa,其他操作条件不变,利用内标法计算得到催化剂活性为21.6×106g/(molcr·h),产物组成记录于表4。
实施例36
将实施例18中的催化剂溶液加入量由10ml改为50ml,其他操作条件不变,利用内标法计算得到催化剂活性为13.5×106g/(molcr·h),产物组成记录于表4。
表4实施例29~36的催化反应结果
由表4可见,活化剂的种类、反应溶剂和催化剂浓度对催化剂的活性以及产物选择性也有较大影响。
- 还没有人留言评论。精彩留言会获得点赞!