一种非均相氮掺杂碳材料负载钴催化剂及维生素K3的生产工艺
一种非均相氮掺杂碳材料负载钴催化剂及维生素k3的生产工艺
技术领域
1.本发明涉及精细化工合成技术领域,具体涉及一种非均相氮掺杂碳材料负载钴催化剂及维生素k3的生产工艺。
背景技术:
2.维生素k3又名甲萘醌,即2-甲基-1,4-萘醌,是一种有机化合物,可衍生制备维生素k1、k2等其他k族维生素。维生素k在临床上属于促凝血药,可以用于治疗维生素k缺乏所引起的出血性疾病,也用于各种食品、饮料和膳食补充剂,有助于人体凝血和保持骨骼健康,对生命健康具有重要意义。维生素k3是工业中主要生产的k族维生素。
3.维生素k3的现有工业生产路线是通过氧化铬或重铬酸钠在醋酸或硫酸中氧化2-甲基萘()实现,但该方法会产生高毒性和致癌性的六价铬,以及大量废料,容易导致环境污染。
4.在实验室研究中,使用较为绿色的双氧水做氧化剂也能实现2-甲基萘到维生素k3的转化,例如,在甲基三氧化铼(mto)催化下,使用双氧水和醋酸酐可以实现58%的产率(angew. chem. int. ed. 1995, 33, 2475-2477),但使用了铼元素,价格较贵;又如,使用精制的氧化钒-氧化铝催化剂(10.1016/j.cattod.2014.12.026),双氧水做氧化剂,也可以实现76%的转化率和54%的选择性,但此反应对催化剂制备过程的配比、煅烧温度要求较高。此外,上述双氧水氧化的方法产率均较低。
5.随着绿色生产的发展趋势要求,采用甲基苯醌作为中间体的生产路线也被提出(参考tetrahedron lett.2010, 51, 2339
–
2341,eur. j. org. chem. 2011, 5355
–
5365),其中涉及三个步骤:对甲酚经过叔丁基过氧化氢氧化得到邻甲基苯醌,邻甲基苯醌与1,3-丁二烯经三氟甲磺酸钪催化的diels-alder反应(狄尔斯-阿尔德反应)得到2-甲基-1,4-四氢萘醌,然后在钯碳催化下脱氢得到维生素k3。该路线中使用高浓度过氧化物来进行甲酚氧化,过氧化物不仅在保存和运输方面存在明显的安全性问题,而且制备成本不低,diels-alder反应使用到了三氟甲磺酸钪,使用了钪元素,价格较贵,同时在2-甲基-1,4-四氢萘醌脱氢的方法中涉及较高成本的钯碳催化剂,不利于工业化大规模应用。
6.中国专利cn111689844a公开了一种由邻甲酚生产2-甲基-1,4-萘醌的新型生产工艺,其在微通道反应器中用50%双氧水氧化邻甲酚,在加压环境下进行diels-alder反应,在溴化铜/溴化锰混合催化剂体系下用dmso(二甲基亚砜)作溶剂和氧化剂来氧化2-甲基-1,4-四氢萘醌。该方法也使用高浓度过氧化物来进行甲酚氧化,不仅在保存和运输方面存在明显的安全性问题,而且制备成本不低;同时diels-alder反应需要使1,3-丁二烯过量添加,且反应使用到了加压环境,条件较为苛刻,存在安全性隐患,耗时较长,经验证在常压下收率低,不利于工业化生产;此外,在2-甲基-1,4-四氢萘醌脱氢的方法中涉及较高成本的
dmso,且生成了二甲基硫醚废弃物,同时催化剂还使用到了卤化物,环保性较差。
技术实现要素:
7.本发明的目的是克服现有技术的一个或多个不足,提供一种新型的可高效高产率地制备2-甲基-1,4-萘醌的催化剂,并且该催化剂易于回收,有利于降低成本。
8.本发明同时还提供了一种2-甲基-1,4-萘醌的制备方法,该方法以特定非均相氮掺杂碳材料负载钴催化剂进行催化脱氢,催化效率高,产率高,环保性好,并且催化剂易于回收,有利于降低成本。
9.为达到上述目的,本发明采用的一种技术方案是:一种非均相氮掺杂碳材料负载钴催化剂,该非均相氮掺杂碳材料负载钴催化剂用于催化2-甲基-1,4-四氢萘醌发生氧化脱氢反应生成2-甲基-1,4-萘醌;该非均相氮掺杂碳材料负载钴催化剂的制备方法包括:将氨基葡萄糖盐酸盐、三聚氰胺和醋酸钴混匀,在惰性气氛下烧结;其中,所述氨基葡萄糖盐酸盐与所述三聚氰胺的投料质量比为1∶25-45,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比为1∶0.01-0.75。
10.本发明提供的又一技术方案:一种非均相氮掺杂碳材料负载钴催化剂在制备2-甲基-1,4-萘醌中的应用,该非均相氮掺杂碳材料负载钴催化剂的制备方法包括:将氨基葡萄糖盐酸盐、三聚氰胺和醋酸钴混匀,在惰性气氛下烧结;其中,所述氨基葡萄糖盐酸盐与所述三聚氰胺的投料质量比为1∶25-45,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比为1∶0.01-0.75。
11.本发明提供的又一技术方案:一种2-甲基-1,4-萘醌的制备方法,该方法包括:使2-甲基-1,4-四氢萘醌在第一氧化剂存在下、在第一催化体系中发生氧化脱氢反应生成2-甲基-1,4-萘醌,反应式为:,其中:所述第一催化体系包含非均相氮掺杂碳材料负载钴催化剂、第一溶剂;该非均相氮掺杂碳材料负载钴催化剂的制备方法包括:将氨基葡萄糖盐酸盐(分子式为:c6h
13
no5·
hcl,分子量为215.63)、三聚氰胺(分子式为c3h6n6,iupac命名为“1,3,5-三嗪-2,4,6-三胺”)和醋酸钴混匀,在惰性气氛下烧结;其中,所述氨基葡萄糖盐酸盐与所述三聚氰胺的投料质量比为1∶25-45,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比为1∶0.01-0.75。
12.根据本发明,本发明方法制备的非均相氮掺杂碳材料负载钴催化剂中,以氮掺杂碳材料作为载体,烧结获得的钴单质作为活性成分被载体氮掺杂碳材料所包覆。
13.在本发明的一些实施方式中,所述惰性气氛可以通过通入氮气、氩气等形成。
14.根据本发明的一些优选方面,所述氨基葡萄糖盐酸盐与所述三聚氰胺的投料质量比为1∶30-40。进一步地,所述氨基葡萄糖盐酸盐与所述三聚氰胺的投料质量比为1∶32-38。
15.根据本发明的一些优选方面,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比
为1∶0.02-0.70。进一步地,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比为1∶0.025-0.45。
16.在本发明的一些优选实施方式中,所述氨基葡萄糖盐酸盐与所述醋酸钴的投料摩尔比为1∶0.1-0.2。
17.根据本发明的一些优选方面,所述烧结在550-850℃下进行。
18.进一步地,所述烧结采用如下方式进行:在550-650℃下烧结,烧结时间为t1;然后按照1.5-3.5℃/min的升温速率升温至750-850℃并烧结,烧结时间为t2,t1、t2分别为0.5-2h。
19.在本发明的一些优选实施方式中,制备所述非均相氮掺杂碳材料负载钴催化剂的实施方式包括:将氨基葡萄糖盐酸盐、三聚氰胺和醋酸钴分散在去离子水中,在加热条件下搅拌直至所述去离子水蒸发完全,然后将干燥的混合物在惰性气氛下烧结,制成所述非均相氮掺杂碳材料负载钴催化剂。
20.根据本发明的一些优选方面,以质量百分含量计,所述非均相氮掺杂碳材料负载钴催化剂的添加质量为所述2-甲基-1,4-四氢萘醌的添加质量的0.1%以上。
21.进一步地,以质量百分含量计,所述非均相氮掺杂碳材料负载钴催化剂的添加质量为所述2-甲基-1,4-四氢萘醌的添加质量的0.5%-10%。更进一步地,以质量百分含量计,所述非均相氮掺杂碳材料负载钴催化剂的添加质量为所述2-甲基-1,4-四氢萘醌的添加质量的0.5%-6%。再进一步地,以质量百分含量计,所述非均相氮掺杂碳材料负载钴催化剂的添加质量为所述2-甲基-1,4-四氢萘醌的添加质量的0.8%-5%。
22.根据本发明,在本发明的催化体系下,所述氧化脱氢反应可以在较低温度下进行。根据本发明的一些优选且具体的方面,所述氧化脱氢反应的反应温度为0-40℃。
23.在本发明的一些实施方式中,所述氧化脱氢反应的反应时间为0.5-5h。
24.在本发明的一些实施方式中,所述氧化脱氢反应的反应压力为0.01-0.15mpa。
25.根据本发明的一些优选方面,所述第一溶剂为醇类溶剂和/或腈类溶剂。
26.根据本发明的一些优选方面,所述醇类溶剂为c1-c6烷基醇。本发明中,“c1-c6烷基醇”是碳原子数为1个、2个、3个、4个、5个或6个的烷基醇。
27.进一步地,所述醇类溶剂为选自甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、正戊醇和异戊醇中的一种或多种的组合。
28.根据本发明的一些优选方面,所述腈类溶剂包含乙腈。
29.根据本发明,在本发明的催化体系下,所述第一氧化剂可以为氧气,绿色环保,可以通过向反应体系中通入纯氧气体或者通入含有氧气的气体混合物(例如空气)以提供所述第一氧化剂。根据本发明,所述纯氧气体中氧气的纯度大于99%,所述含有氧气的气体混合物中氧气浓度低于99%。
30.在本发明的一些优选实施方式中,所述氧化脱氢反应实施如下:将2-甲基-1,4-四氢萘醌、非均相氮掺杂碳材料负载钴催化剂、第一溶剂均匀混合,然后通入纯度大于99%的纯氧气体或空气,控制反应温度为0-40℃、反应压力为0.01-0.15mpa,进行氧化脱氢反应,生成2-甲基-1,4-萘醌。
31.根据本发明的一些优选方面,所述2-甲基-1,4-萘醌的制备方法还包括制备2-甲基-1,4-四氢萘醌的步骤,该制备2-甲基-1,4-四氢萘醌的步骤包括:
使邻甲基苯醌与1,3-丁二烯在式(ⅰ)所示的金属螯合型离子液体催化作用下发生加成反应,生成2-甲基-1,4-四氢萘醌,反应式为:;(ⅰ),a、b、c、d分别独立地为0、1或2。本发明中,该式(ⅰ)所示的金属螯合型离子液体作为路易斯酸催化邻甲基苯醌与1,3-丁二烯的diels-alder反应,效果显著。另外该金属螯合型离子液体不仅有很好的热稳定性,在常规有机溶剂及水中都有非常好的溶解度,因此可以通过精馏的方法回用催化剂,也可以通过水洗分离的方法回用催化剂。
32.根据本发明的一些优选且具体的方面,a、b、c和d的取值相同。
33.根据本发明的一个具体方面,式(ⅰ)所示的金属螯合型离子液体(chelate-cu-il)结构如下所示:。
34.根据本发明的一些优选方面,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶5-5000。
35.进一步地,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶5-2500。
36.更进一步地,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶5-2000。
37.再进一步地,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶5-1000。
38.在本发明的一些优选且具体的实施方式中,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶5-500。
39.在本发明的一些实施方式中,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶10-5000。进一步地,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶10-2500。更进一步地,所述式(ⅰ)所示的金属螯合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶10-2000。再进一步地,所述式(ⅰ)所示的金属螯
合型离子液体与所述邻甲基苯醌的投料摩尔比为1∶10-1000。
40.根据本发明,在本发明金属螯合型离子液体的催化作用下,所述加成反应可以在较低温度下进行;在本发明的一些实施方式中,所述加成反应的反应温度为0-80℃,进一步地,可以在0-70℃下进行。
41.根据本发明,在本发明金属螯合型离子液体的催化作用下,所述加成反应的反应压力可以为常压,无需加压处理,在常压下进行即可获得理想的产物收率。
42.根据本发明,在本发明金属螯合型离子液体的催化作用下,邻甲基苯醌与1,3-丁二烯的投料摩尔比可以为1∶0.95-1.05。根据本发明的一个具体方面,邻甲基苯醌与1,3-丁二烯的投料摩尔比可以为1∶1。
43.根据本发明的一些优选方面,所述式(ⅰ)所示的金属螯合型离子液体的制备方法包括:使式(ⅱ)所示化合物与式(ⅲ)所示化合物在第二溶剂中反应制成式(ⅰ)所示的金属螯合型离子液体:。
44.式(ⅲ)中,a、b、c和d的定义同前。
45.根据本发明的一些优选且具体的方面,所述第二溶剂包含丙酮。在本发明的一些实施方式中,所述第二溶剂为丙酮。
46.根据本发明的一些优选且具体的方面,制备所述式(ⅰ)所示的金属螯合型离子液体的过程中,所述反应在10-40℃下进行。
47.根据本发明的一些优选方面,所述式(ⅱ)所示化合物采用如下路线合成:,其中,使2,6-二甲基苯胺与2,6-吡啶二甲酰氯反应生成式(ⅳ)所示化合物,使式(ⅳ)所示化合物、氯化铜和甲醇反应生成式(ⅱ)所示化合物。
48.根据本发明的一些优选方面,在制备式(ⅳ)所示化合物的过程中,使反应在碱性环境下进行,所述碱性环境通过添加碱性试剂形成,所述碱性试剂包含三乙胺。
49.根据本发明的一些优选方面,在制备式(ⅳ)所示化合物的过程中,使反应在10-40℃、在第三溶剂中进行,所述第三溶剂包含卤代c1-c3烷基。在本发明的一些实施方式中,所述卤代c1-c3烷基中的卤素为选自氟、氯和溴中的一个或多个,进一步地,所述卤代c1-c3烷基可以为二氯甲烷、二氯乙烷等。
50.根据本发明的一些优选方面,在制备式(ⅱ)所示化合物的过程中,使式(ⅳ)所示化合物、氯化铜和甲醇的反应在10-40℃、碱金属的甲醇盐存在下进行,所述碱金属的甲醇
盐为选自甲醇钠、甲醇钾和甲醇锂中的一种或多种的组合。
51.根据本发明的一些优选方面,所述2-甲基-1,4-萘醌的制备方法还包括制备邻甲基苯醌的步骤,该制备邻甲基苯醌的步骤包括:使邻甲酚和/或间甲酚在第二催化体系中、在第二氧化剂存在下发生氧化反应,生成邻甲基苯醌,反应式为:;其中,所述第二氧化剂为氧气;所述第二催化体系包含式(
ⅴ
)所示的4-r-2,2,6,6-四甲基哌啶氧化物、亚硝酸盐、质子酸和第四溶剂,所述第四溶剂为选自甲醇、乙醇、丙醇、叔丁醇、水和乙腈中的一种或多种的组合;(
ⅴ
),r为-h、-oh、-nhac、-cooh、-cooph或。本发明中,在本发明第二催化体系作用下,可高收率的获得目的产物,尤其是可以采用氧气作为氧化剂,且不需要使用过渡金属等催化剂,成本低廉,绿色环保;本发明发现只有在亚硝酸盐和质子酸协同作用下,以及在上述特定的溶剂例如甲醇、乙醇、丙醇、叔丁醇、水或乙腈中,才能获得较好的收率。同时本发明在无金属催化剂的条件下,使用有机催化剂4-r-2,2,6,6-四甲基哌啶氧化物(4-r-tempo)实现了邻甲酚到邻甲基苯醌的氧气氧化,可避免卤素离子及过渡金属的使用,有利于工业生产及维生素k3药品的品质提高。
52.根据本发明的一些优选且具体的方面,制备邻甲基苯醌的过程中,在所述氧化反应过程中,通过向反应体系中纯氧气体或者通入含有氧气的气体混合物(例如空气)以提供所述第二氧化剂。
53.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述氧化反应的反应温度为20-80℃。
54.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述氧化反应的反应压力为0.1-20mpa。
55.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述邻甲酚和/或间甲酚、所述式(
ⅴ
)所示的4-r-2,2,6,6-四甲基哌啶氧化物、所述亚硝酸盐和所述质子酸的投料摩尔比为10-200∶1∶1-4∶1-4。
56.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述式(
ⅴ
)所示的4-r-2,2,6,6-四甲基哌啶氧化物、所述亚硝酸盐和所述质子酸的总添加量与所述邻甲酚和/或间甲酚的投料摩尔比为0.01-1∶1。
57.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述质子酸为选自三氟甲磺酸、甲磺酸、硫酸、对甲苯磺酸和氯化氢醇溶液中的一种或多种的组合。在本发明的一些实施方式中,氯化氢醇溶液可以采用添加氯化氢-乙醇溶液的形式加入。
58.进一步优选地,所述质子酸为三氟甲磺酸和/或甲磺酸。
59.根据本发明的一些优选方面,制备邻甲基苯醌的过程中,所述亚硝酸盐为亚硝酸钾和/或亚硝酸钠。
60.根据本发明的一些优选方面,该制备邻甲基苯醌的步骤中,r为-h、-oh、-nhac、-cooh、-cooph或,所述质子酸为三氟甲磺酸、甲磺酸中的至少一种,所述第四溶剂为甲醇、叔丁醇、乙腈中的至少一种,所述邻甲酚和/或间甲酚、所述式(ⅱ)所示的4-r-2,2,6,6-四甲基哌啶氧化物、所述亚硝酸盐和所述质子酸的投料摩尔比为10-50∶1∶1.5-2.5∶1.5-2.5。
61.本发明中,醋酸钴可以带有结晶水或经处理不含结晶水。
62.本发明中,“第一”、“第二”、“第三”、“第四”的描述仅是为了区分各个反应中所用到的催化体系或者溶剂等,便于描述,不易混淆,不代表先后关系,也不意味着相同或者不同。
63.由于上述技术方案运用,本发明与现有技术相比具有下列优点:采用本发明的非均相氮掺杂碳材料负载钴催化剂能够实现由2-甲基-1,4-四氢萘醌氧化脱氢高效高选择性地制备2-甲基-1,4-萘醌,该过程兼具环保性好,并且催化剂易于回收套用,有利于降低成本。
附图说明
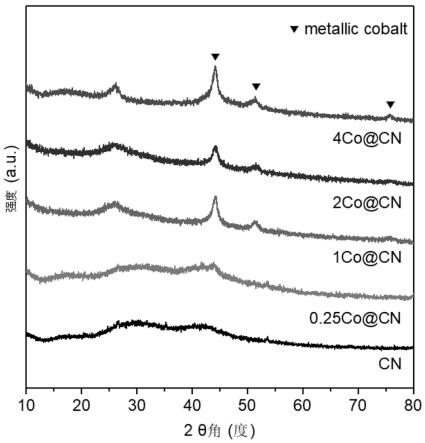
64.图1为不同钴含量的co@cn催化剂的x射线衍射谱图,实施例1使用的是1co@cn催化剂,实施例8使用的是0.25co@cn,实施例10使用的是使用的是2co@cn,实施例11使用的是4co@cn催化剂;图2为本发明实施例1中1co@cn催化剂与cn的x射线光电子能谱图(氧1s电子);图3为本发明实施例1中1co@cn催化剂与cn的x射线光电子能谱图(氮1s电子);图4为本发明实施例1中1co@cn催化剂不同结合能下的x射线光电子能谱图(钴2p电子);图5为本发明实施例1的1co@cn催化剂与对比例5的co/ac催化剂的x射线衍射谱图;图6为本发明实施例13所制备的铜螯合型离子液体chelate-cu-il中阳离子的电喷雾高分辨质谱图、原子吸收表征图谱;图7为本发明实施例13所制备的铜螯合型离子液体chelate-cu-il中阴离子的电喷雾高分辨质谱图、原子吸收表征图谱。
具体实施方式
65.以下结合具体实施例对上述方案做进一步说明;应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
66.下述实施例中未作特殊说明,所有原料均来自于商购或通过本领域的常规方法制备而得。
67.下述中,所有反应转化率和产率使用气相色谱测定(使用商业渠道购买的原料和产物定标准曲线)。
68.下述中,非均相氮掺杂碳材料负载钴催化剂yco@cn的制备方法:将 2g gah(氨基葡萄糖盐酸盐)、70g三聚氰胺和y mmol醋酸钴(由于原料醋酸钴易潮解带有水,则该醋酸钴可采用水合物形式添加,通常添加四水合乙酸钴,也可以脱水后再添加,仅需控制醋酸钴的摩尔添加量即可)的混合物溶解在去离子水中,然后在80℃下搅拌以蒸发溶液。将干燥的固体转移到管式炉中,并在590℃下在氮气气氛中煅烧1小时,然后以2.5℃/min的升温速率加热至790℃,并在790℃下保持50分钟,氮气流速为400ml/min,自然冷却,得到催化剂,记为yco@cn。
实施例1
69.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该方法采用如下合成路线: ;该方法包括如下步骤:在20ml的反应瓶中,加入3mmol 2-甲基-1,4-四氢萘醌、0.01g的1co@cn、4.0g甲醇,加入磁子,接好氧气气囊(0.1mpa,纯度大于99%的纯氧气体),换气3次,在25℃下反应1小时,过滤回收催化剂,并用溶剂洗涤催化剂3次,合并母液进行上塔精馏,得到纯品。反应转化率为99.3%,维生素k3产率为98.1%,纯度为98.7%;其中,1co@cn催化剂的制备:将 2g gah、70g三聚氰胺和1mmol醋酸钴的混合物溶解在去离子水中,然后在80℃下搅拌以蒸发溶液。将干燥的固体转移到管式炉中,并在590℃下在氮气气氛中煅烧1小时,然后以2.5℃/min的升温速率加热至790℃,并在790℃下保持50分钟,氮气流速为400ml/min,自然冷却,得到催化剂,记为1co@cn,其x射线衍射谱图(xrd)及x射线光电子能谱图(xps)参见图1-图4。
实施例2
70.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂为0.004g 1co@cn,其余与实施例1完全相同,反应转化率为92.8%,维生
素k3产率为89.4%,纯度为96.1%。
实施例3
71.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂为0.02g 1co@cn,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为98.7%,纯度为99.3%。
实施例4
72.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂为0.005g 1co@cn,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为94.2%,纯度为98.9%。
实施例5
73.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,将溶剂甲醇替换为丙醇,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为96.4%,纯度为99%。
实施例6
74.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,反应温度由25℃调整为0℃,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为91.1%,纯度为96.8%。
实施例7
75.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,反应温度由25℃调整为40℃,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为92.6%,纯度为98.7%。
实施例8
76.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂的制备过程中醋酸钴添加量为0.25mmol,制备的催化剂记为0.25co@cn,x射线衍射谱图(xrd)参见图1,其余与实施例1完全相同,反应转化率为92.2%,维生素k3产率为90.6%,纯度为97.7%。
实施例9
77.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂的制备过程中醋酸钴添加量为1.5mmol,其余与实施例1完全相同,反应转化率为100%,维生素k3产率为98.9%,纯度为99%。
实施例10
78.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂的制备过程中醋酸钴添加量为2mmol,制成的催化剂记为2co@cn,x射线衍射谱图(xrd)参见图1,其余与实施例1完全相同,反应转化率为96.3%,维生素k3产率为92.7%,纯度为97.8%。
实施例11
79.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例1相比,区别之处在于,催化剂的制备过程中醋酸钴添加量为4mmol,催化剂记为4co@cn,x射线衍射谱图(xrd)参见图1,其余与实施例1完全相同,反应转化率为94%,维生素k3产率为91.3%,纯度为97.5%。
实施例12 催化剂回收套用实验
80.回收套用催化剂,我们对2-甲基-1,4-四氢萘醌氧化反应进行了放大实验,具体步骤如下:在100ml的反应瓶中,加入30mmol 2-甲基-1,4-四氢萘醌、0.1g的1co@cn、40g甲醇,加入磁子,接好氧气气囊(0.1mpa,纯度大于99%的纯氧气体),换气3次,在25℃下反应1小时,过滤回收催化剂,并用溶剂洗涤催化剂3次,合并母液进行上塔精馏,得到纯品。反应转化率为100%,维生素k3产率为98.5%,纯度为99%,说明放大10倍对收率和产品纯度没有明显影响。
81.催化剂co@cn回收方法:将反应液过滤分离co@cn催化剂,分别用适量丙酮和甲醇浸洗co@cn催化剂三次,过滤,然后放于真空干燥箱中,60℃干燥8 h,再进行套用。
82.进一步地,与实施例1相比,区别之处在于,使用的1co@cn催化剂是回收的催化剂,其余与实施例1完全相同,反应转化率为99.0%,维生素k3产率为97.2%,纯度为99.1%。产率没有明显降低,这说明1co@cn催化剂具有较好的稳定性。此外,再次进行套用实验,反应转化率为98.4%,维生素k3产率为96.3%,纯度为98.1%,转化率和产率降低了一些,这可能是由于在回收过程中过滤洗涤操作导致催化剂质量减少引起的。
对比例1
83.该例与实施例1相比,区别之处在于,催化剂制备过程中醋酸钴的添加量为9mmol,其余与实施例1完全相同,反应转化率为85.8%,维生素k3产率为90.7%,纯度为97.4%。
对比例2
84.本例与实施例1相比,区别之处在于,1co@cn制备过程中用硝酸钴(带有6个结晶水,即六水合硝酸钴)代替醋酸钴,六水合硝酸钴的添加量为1mmol,其余与实施例1完全相同,反应转化率为62.1%,维生素k3产率为54.1%,纯度为83.6%。这说明不同钴盐对催化性能影响较大。
对比例3
85.本例与实施例1相比,区别之处在于,催化剂替换为co3o4,其余与实施例1完全相
四甲基哌啶氧化物的结构式为:;4-maleimide(马来酰亚胺基)-2,2,6,6-四甲基哌啶氧化物的结构式为:;4-nh
2-2,2,6,6-四甲基哌啶氧化物的结构式为:。
91.下述中,三氟甲磺酸、对甲苯磺酸、甲磺酸分别采用市售产品,纯度:99%;醋酸采用市售产品,纯度:99.5%;硝酸采用质量浓度为98%的发烟硝酸,分析纯试剂;硫酸采用市售产品,纯度:ar(沪试),95.0~98.0%;氯化氢采用市售产品氯化氢-乙醇溶液,氯化氢的浓度为2mol/l;各个酸的投料量根据所需摩尔量换算得到。
实施例13
92.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该方法采用如下合成路线:;具体步骤包括:(1)邻甲基苯醌的制备:在20ml的反应瓶中加入1mmol邻甲酚、0.05mmol 4-oh-tempo、0.1mmol亚硝酸钠、0.1mmol三氟甲磺酸以及2ml甲醇,加入磁子,通氧气(纯度大于99%的纯氧气体),压力0.5mpa,在30℃、800rpm搅拌下反应18小时,反应转化率为100%,邻甲基苯醌产率为92.7%;反应液旋去溶剂后溶于10ml乙酸乙酯中,用2ml氢氧化钠水溶液洗两次,用5ml饱和食盐水萃洗3次,有机相用无水硫酸钠干燥后旋干,得到纯化的邻甲基苯醌,纯度99%。
93.(2)2-甲基-1,4-四氢萘醌的制备:在20ml的反应瓶中加入0.2mmol chelate-cu-il、2mmol按照上述方法制备的邻甲基苯醌与2mmol 1,3-丁二烯,70℃反应8小时,反应转化率为100%,2-甲基-1,4-四氢萘醌产率为95.1%;反应液旋去溶剂后溶于10ml乙酸乙酯中,用5ml饱和食盐水萃洗3次,有机相用
无水硫酸钠干燥后旋干,得到纯化的2-甲基-1,4-四氢萘醌,纯度为98%;其中,铜螯合型离子液体chelate-cu-il(式(ⅰ))采用如下方法制备:反应式如下:;具体地:在250ml圆底烧瓶中加入2.6-二甲基苯胺(10mmol)和三乙胺(26mmol),在0℃下,缓慢向烧瓶中滴加2,6-吡啶二甲酰氯(5mmol)的二氯甲烷(20ml)混合液;室温搅拌24h后,过滤除去溶剂,滤饼用蒸馏水和乙醚洗涤3-5次,40℃真空干燥得到白色粉末配体(式(ⅳ)所示化合物)。将配体(2.0mmol)、无水氯化铜(2.0mmol)和甲醇(100ml)加入到250ml圆底烧瓶中,在混合液中加入浓度为0.5mol/l甲醇钠的甲醇溶液 (8.0ml);室温搅拌反应24h,40℃旋蒸得到油状固体;将固体用乙腈(20ml)溶解,并加入甲苯(100ml)静止30min后过滤,滤液40℃真空旋蒸,得到绿色粉末(式(ⅱ)所示化合物),真空干燥过夜。在50ml 圆底烧瓶中加入绿色粉末(式(ⅱ)所示化合物)和三己基(十四烷基)氯化磷(式(
ⅲ‑
1)所示化合物)各2mmol,加入10ml丙酮,室温搅拌12h,旋蒸得到绿色液体后,用二氯甲烷(3
×
5ml)萃取,下层有机相再次40℃真空旋蒸除去溶剂后,真空干燥24h,即得到产物:铜螯合型离子液体chelate-cu-il(式(ⅰ));铜螯合型离子液体chelate-cu-il(式(ⅰ))的元素分析表参见表1:
94.其电喷雾高分辨质谱图与原子吸收表征图谱具体参见图6和图7所示;2-甲基-1,4-萘醌(维生素k3)的制备:采用实施例1的方法进行。
实施例14
95.本例提供一种2-甲基-1,4-四氢萘醌(维生素k3中间体)的制备方法,该例与实施
例13相比,区别之处在于,步骤(1)中底物为1mmol间甲酚,其余与实施例13完全相同,反应转化率为98.2%,邻甲基苯醌产率为87.9%,纯度为97%。
实施例15
96.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.005mmol 4-nhac-2,2,6,6-四甲基哌啶氧化物,0.01mmol三氟甲磺酸,0.01mmol nano2,其余与实施例13完全相同,反应转化率为89.4%,邻甲基苯醌产率为80.1%,纯度为95%。
实施例16
97.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.1mmol 4-nhac-2,2,6,6-四甲基哌啶氧化物,0.2mmol三氟甲磺酸,0.2mmol nano2,其余与实施例13完全相同,反应转化率为100%,邻甲基苯醌产率为92.4%,纯度为98%。
实施例17
98.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中通空气,其余与实施例13完全相同,反应转化率为100%,邻甲基苯醌产率为86.4%,纯度为97%。
实施例18
99.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中将溶剂甲醇调整为2 ml乙腈,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为89.2%,纯度为97%。
实施例19
100.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中将溶剂甲醇调整为2 ml叔丁醇,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为92.3%,纯度为97%。
实施例20
101.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中将溶剂甲醇调整为1.6ml叔丁醇加0.4ml乙腈,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为94.3%,纯度为98%。
实施例21
102.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05mmol 4-cooh-2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为88.5%,纯度为95%。
实施例22
103.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05mmol 4-nhac-2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为89.7%,纯度为97%。
实施例23
104.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05mmol 2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为90.6%,纯度为98%。
实施例24
105.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05 mmol 4-maleimide(马来酰亚胺基)-2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为91.6%,纯度为99%。
实施例25
106.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05 mmol 4-cooph-2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为89.9%,纯度为97%。
实施例26
107.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中质子酸为0.1 mmol 甲磺酸,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为93.1%,纯度为99%。
实施例27
108.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中质子酸为0.1 mmol h2so4,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为81.4%,纯度为95%。
实施例28
109.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中质子酸为0.1 mmol hcl,其余与实施例13完全相同,转化率为100%,邻甲基苯醌产率为79.4%,纯度为96%。
实施例29
110.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中质子酸为0.1 mmol 对甲苯磺酸,其余与实施例13完全相同,转化率
为89.2%,邻甲基苯醌产率为74.1%,纯度为98%。
实施例30
111.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中三氟甲磺酸为0.2 mmol,nano2为0.2 mmol,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为84.9%,纯度为96%。
实施例31
112.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中三氟甲磺酸为0.05 mmol,nano2为0.05 mmol,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为79.5%,纯度为93%。
实施例32
113.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中反应温度为20℃,压力20mpa,其余与实施例13完全相同,转化率为95.4%, 邻甲基苯醌产率为84.6%,纯度为96%。
实施例33
114.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中反应温度为80℃,压力为0.1mpa,其余与实施例13完全相同,转化率为93.1%, 邻甲基苯醌产率为83.3%,纯度为94%。
实施例34
115.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(1)中反应温度为40℃,压力为10mpa,其余与实施例13完全相同,转化率为100%, 邻甲基苯醌产率为90.4%,纯度为97%。
实施例35
116.本例提供一种2-甲基-1,4-四氢萘醌(维生素k3中间体)的制备方法,该例与实施例13相比,区别之处在于,步骤(2)中,催化剂为0.0004mmol chelate-cu-il,制备2-甲基-1,4-四氢萘醌的反应温度为80℃,其余与实施例13完全相同,反应转化率为92.2%,2-甲基-1,4-四氢萘醌产率为81.5%,纯度为91%。
实施例36
117.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(2)中,催化剂为0.002mmol chelate-cu-il,制备2-甲基-1,4-四氢萘醌的反应温度为40℃,其余与实施例13完全相同,反应转化率为100%,2-甲基-1,4-四氢萘醌产率为92.5%,纯度为97%。
实施例37
118.本例提供一种2-甲基-1,4-萘醌(维生素k3)的制备方法,该例与实施例13相比,区别之处在于,步骤(2)中,催化剂为0.04mmol chelate-cu-il,制备2-甲基-1,4-四氢萘醌的反应温度为0℃,其余与实施例13完全相同,反应转化率为90.9%, 2-甲基-1,4-四氢萘醌产率为89.4%,纯度为95%。
对比例6
119.该例与实施例13相比,区别之处在于,步骤(1)中催化剂为0.05mmol 4-nh
2-2,2,6,6-四甲基哌啶氧化物,其余与实施例13完全相同,转化率为80.9%,邻甲基苯醌产率为69.3%,纯度为86%。
对比例7
120.该例与实施例13相比,区别之处在于,步骤(1)中将三氟甲磺酸替换为0.1mmol醋酸,其余与实施例13完全相同,转化率为50.9%, 邻甲基苯醌产率为41.4%,纯度为90%。
对比例8
121.该例与实施例13相比,区别之处在于,步骤(1)中将三氟甲磺酸替换为0.1mmol硝酸,其余与实施例13完全相同,转化率为85.4%, 邻甲基苯醌产率为50.2%,纯度为86%。
对比例9
122.该例与实施例13相比,区别之处在于,步骤(1)中反应过程中未加入三氟甲磺酸,其余与实施例13完全相同,反应转化率为13.1%,邻甲基苯醌产率为12.1%。
123.对比例7-9说明三氟甲磺酸、甲磺酸、对甲苯磺酸类质子酸是歧化tempo催化剂,起到催化作用的关键所在,且是高收率获得的关键因素之一。
对比例10
124.该例与实施例13相比,区别之处在于,步骤(1)中将溶剂2ml甲醇替换为2ml二甲基亚砜(dmso),其余与实施例13完全相同,反应转化率为20.8%,邻甲基苯醌产率为2.1%。
对比例11
125.该例与实施例13相比,区别之处在于,步骤(1)中将溶剂2ml甲醇替换为2ml二氯甲烷,其余与实施例13完全相同,反应转化率为96.4%,邻甲基苯醌产率为34.3%。
对比例12
126.该例与实施例13相比,区别之处在于,步骤(1)中将溶剂2ml甲醇替换为2ml氯苯,其余与实施例13完全相同,反应转化率为100%,邻甲基苯醌产率为23.1%。
127.该对比例10-12说明本发明特定的溶剂体系对邻甲基苯醌的产率有决定性的作用。
对比例13
128.该例与实施例13相比,区别之处在于,步骤(1)中反应过程中未加入任何tempo催化剂,其余与实施例13完全相同,反应转化率为0%,邻甲基苯醌产率为0%。该对比例说明tempo催化剂对活化邻甲酚,并使其转化为邻甲基苯醌有关键作用。
对比例14
129.该例与实施例13相比,区别之处在于,步骤(1)中未加入nano2,其余与实施例13完全相同,反应转化率为8.4%,邻甲基苯醌产率为7.1%。该对比例说明亚硝酸盐起到循环tempo催化剂,从而完全转化底物的作用。
对比例15
130.该例与实施例13相比,区别之处在于,步骤(2)中,将铜螯合型离子液体chelate-cu-il替换为“chelate-cu-meoh”,其余与实施例13完全相同,反应转化率为63.5%,2-甲基-1,4-四氢萘醌产率为55.7%,纯度为82%。
131.。
对比例16
132.该例与实施例13相比,区别之处在于,步骤(2)中,不加铜螯合型离子液体chelate-cu-il,其余与实施例13完全相同,反应转化率为36.9%,邻甲基苯醌产率为30.1%,纯度为76%。
133.上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
134.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1