使用包括含有沸石和加氢金属的富铈稀土的催化剂的烷基化工艺的制作方法
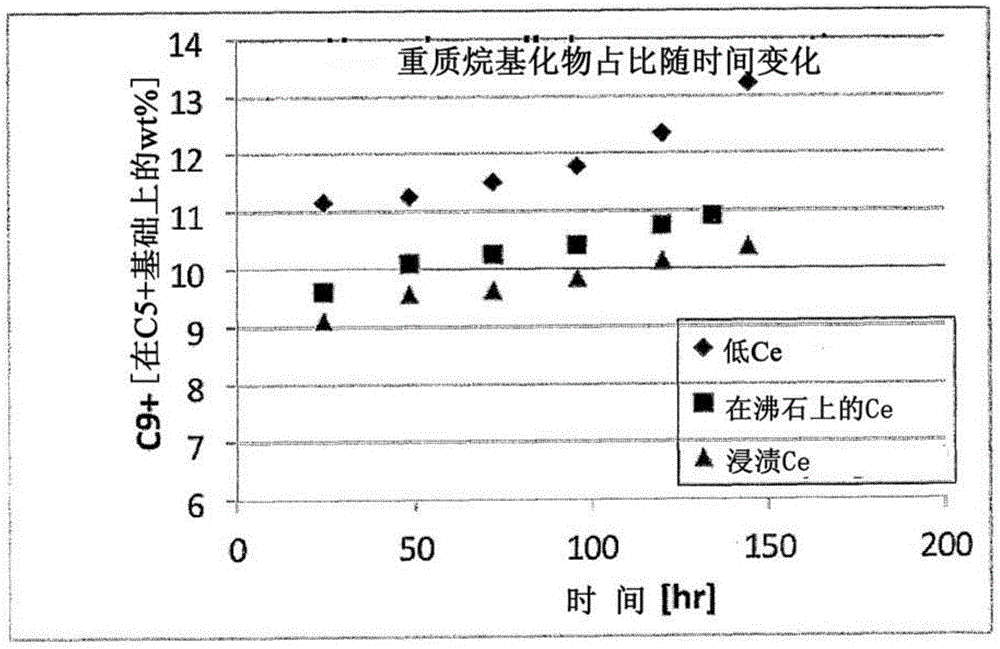
烷基化这个术语指的是可烷基化化合物例如芳香族或饱和烃,与烷化剂例如烯烃之间的反应。反应是令人感到有兴趣的因为它使得有可能通过异链烷烃例如异丁烷与含有2-6个碳原子的烯烃的烷基化,得到具有高辛烷值且在汽油馏分范围内的烷基化物。有别于通过裂解重质石油馏分得到的汽油例如真空瓦斯油和常温渣油,烷基化得到的汽油基本不含有例如硫和氮等污染物且因此具有清洁燃烧的特点。由高辛烷值表示的其高抗爆性减少了添加对环境有害的抗爆化合物的需要,例如芳烃或铅。也有别于重整石脑油或裂解重质石油馏分得到的汽油,烷基化物即使含有芳烃或烯烃,也不多,其提供了进一步的环保优势。烷基化反应是酸催化的。传统的烷基化工艺设备使用液体酸催化剂例如硫酸和氢氟酸。这种液体酸催化剂的使用带来了各种各样的问题。例如,硫酸和氢氟酸都是强腐蚀性的,使得使用的设备必须要满足苛刻的服务要求。因为在生成的燃料中强腐蚀性材料的存在是有害的,残留的酸必须从烷基化物中被除去。也因为必须要进行的液相分离,工艺是复杂和昂贵的。此外,常常具有风险:有毒物质例如氢氟酸将被排放到环境中。历史上相比有竞争性的液体酸烷基化工艺,固体酸烷基化催化剂的活性和稳定性仍然具有需要改进的地方。固体酸烷基化催化剂中最近的进展包括:如在WO/9823560(美国专利5,986,158号)中公开的采用含沸石易再生的固体酸烷基化催化剂的烷基化工艺,根据美国专利申请公开2007/0293390号的改进的固体酸催化剂生产工艺,根据WO2005/075387的烷基化催化剂水化工艺,根据美国专利7,176,340号,US2002/198422和EP1485334的连续或半连续烷基化和再生过程,以及在美国专利申请公开2008/0183025号中指出的稀土(RE)交换固体酸催化剂。生成活跃和稳定的固体酸烷基化催化剂的另一个历史上的尝试包括美国专利3,851,004号。‘004参考文献涉及使用含沸石催化剂烃类的烷基化工艺且尤其是芳香族或异链烷烃烷基化工艺,其中反应是通过沸石分子筛催化剂连同第VIII族金属加氢剂催化。然而,‘004参考文献专门指出稀土阳离子的加入是不重要的。生成活跃和稳定的固体酸烷基化催化剂的其它现有技术尝试包括美国专利8,163,969号,美国专利申请公开2010/0234661号和美国专利申请公开2011/0313227号。这里包括的这些参考文献仅供参考。这些现有技术尝试公开了以这种固体酸烷基化催化剂的稀土交换分子筛(例如Y型沸石)。仍然需要一种稳定和活跃的固体酸催化剂。本发明提供一种改进的烷基化工艺,使用包括含有沸石和加氢金属的富铈稀土的固体酸催化剂。技术实现要素:已发现:相比低铈稀土,在这种固体酸烷基化催化剂中对含有交换分子筛(例如Y型沸石)的富铈稀土的使用提高了催化剂的烷基化活性和稳定性。在本发明的一个实施例中提供了包括加氢金属和以含有分子筛的富铈稀土的形式的固体酸的固体催化剂,其中催化剂的特征是至少孔直径在100nm以下的孔的孔隙度小于0.20ml/g,,且总孔隙度大于0.30ml/g。在本发明的另一个实施例中提供了烃的烷基化工艺,包括在烷基化工艺条件下饱和烃原料和一种或多种烯烃与这个发明的催化剂接触。这些以及更进一步的实施例,本发明的特点和优势将通过下文包括附图和权利要求的详细描述的更加清楚。附图说明图1为烯烃转化率随时间曲线图;图2为C9+化合物产率随时间曲线图;图3为烯烃转化率随时间曲线图;图4为C9+化合物产率随时间曲线图。具体实施方式所提到的有关催化剂组份的所有重量百分比基于干燥催化剂(在600℃加热1小时)。稀土的wt%以干燥基(600℃,1小时)稀土氧化物计算。催化剂的含水量范围从大约1.5wt%至大约6wt%,在一个实施例中范围从大约1.8wt%至大约4wt%,且在另一个实施例中范围从大约2wt%至大约3wt%。催化剂的含水量被定义为在烷基化工艺中使用期间的含水量且通过加热催化剂至600℃包括在600℃保持两小时后确定的水份损失而测定(LOI600)。催化剂进一步包括加氢金属。合适的加氢金属实例是过渡金属,例如元素周期表中第VIII族的金属及其混合物。在这些中,元素周期表中第VIII族的贵金属是优选的。铂、钯以及其混合物是尤其优选的。加氢金属的数量将根据其性质而定。当加氢金属是元素周期表中第VIII族的贵金属时,催化剂通常含有范围为大约0.01至大约2wt%的金属。在一个实施例中范围从大约0.1至大约1wt%,以金属计算且基于催化剂总重量。催化剂进一步包括固体酸。固体酸的实例为沸石例如β-沸石,MCM-22,MCM-36,丝光沸石,八面沸石例如X型沸石和Y型沸石,包括H-Y-沸石和USY-沸石,非沸石固体酸例如二氧化硅-氧化铝,硫酸化氧化物例如锆、钛、或锡的硫酸化氧化物,锆、钼、钨、磷等的混合氧化物,和氯化铝氧化物或粘土。优选的固体酸为沸石,包括丝光沸石、β-沸石,八面沸石例如X型沸石和Y型沸石,包括H-Y-沸石和USY-沸石。固体酸的混合物也可被使用。在一个实施例中固体酸为八面沸石其晶胞常数(a0)为24.72至大约25.00埃,在另一个实施例中固体酸是Y型沸石其晶胞常数为24.34-24.72埃,而在另一个实施例中固体酸为Y型沸石其晶胞常数为24.42-24.56埃。在另一个实施例中固体酸为Y型沸石其晶胞常数为24.56-24.72埃。催化剂包括稀土,即选自镧系元素的元素或这些元素的组合。在一个实施例中,催化剂的固体酸组份包括大约0.5wt%至大约32wt%的稀土。在另一个实施例中,催化剂的固体酸组份包括大约2wt%至大约9wt%的稀土。在另一个实施例中,催化剂的固体酸组份包括大约4wt%至大约6wt%的稀土。此外,催化剂的稀土元素组份至少部分为铈。在最终的催化剂中铈的含量应当为多于0.1wt%。更优选的,铈含量应当为多于0.3wt%。最优选的,铈含量应当为至少0.5wt%。稀土元素可通过常规方式被交换到固体酸组份中。在一个实施例中,固体酸组份中的稀土元素基本全是铈。在另一个实施例中,固体酸组份中的稀土元素是富铈稀土混合物。在这个混合物中,铈的含量应当为多于混合物的3wt%。更优选的,铈含量应当为多于混合物的5wt%。最优选的,铈含量应当至少为混合物的10wt%。其余稀土混合物将基本包括一种或多种其它稀土元素,即选自镧系的元素例如镧或这些元素的组合。在另一个实施例中,额外的铈被添加到催化剂中。这通过含固体酸粒子的浸渍和/或离子交换进行。例如,这个工艺可通过孔体积浸渍实现,使用硝酸铈或氯化铈溶液,且相比于催化剂的水孔体积大约95-115wt%,优选的105wt%饱和度。接着,在大约在380-550℃优选的420-500℃煅烧。优选的,煅烧之前,催化剂是干燥的,优选的在大约110-150℃干燥,更优选的在120-130℃。或者,催化剂粒子用铈溶液交换且与在浸渍后使用的相似条件下干燥和煅烧。优选的,在向催化剂加入第VIII族金属之前,用铈进行离子交换和/或浸渍。在固体酸组份的交换工艺期间,钠(Na+)从催化剂中被除去。在一个实施例中固体酸组份含有少于1.5wt%的Na2O。在另一个实施例中,少于1.0wt%的Na2O。在另一个实施例中,少于0.6wt%的Na2O,所有的都以干燥基计算(600℃,1小时)。催化剂可另外包括基质材料。适合的基质材料的例子为氧化铝,二氧化硅,二氧化钛,氧化锆,粘土及其混合物。包括氧化铝的基质材料通常是优选的。在一个实施例中,催化剂包括大约2wt%至大约98wt%的固体酸和大约98wt%至大约2wt%的基质材料,基于催化剂中存在的固体酸和基质材料的总重量。在另一个实施例中,催化剂包括大约10wt%至大约90wt%的固体酸和大约90wt%至大约10wt%的基质材料,基于催化剂中含有的固体酸和基质材料的总重量。在另一个实施例中,催化剂包括大约10wt%至大约80wt%的基质材料和其余的固体酸。在另一个实施例中,催化剂包括大约10wt%至大约40wt%的基质材料和其余的固体酸,基于催化剂中含有的固体酸和基质材料的总重量。催化剂优选的含有少于0.5%wt的卤素。更优选的催化剂含有仅仅是微量卤素。生成的催化剂的孔直径低于100nm的孔的孔体积以及总孔体积通过基于沃什伯恩(Washburn)公式的压汞(Hg)法测定,D为孔直径,p为测量期间施加的压力,γ为表面张力,为480dynes/cm,以及θ为接触角,为140°。在目前的测量中,压力在使得测量覆盖的孔直径范围为4.2-8000nm的范围内变化。在一个实施例中,催化剂具有总孔体积至少为大约0.23ml/g和在另一个实施例中至少大约0.25ml/g。更优选的,总孔体积至少为0.3ml/g,和最优选的是至少0.4ml/g。催化剂粒子可具有多种不同形状,包括球形,圆柱形,环形和对称或不对称多叶形例如三叶和四叶。在一个实施例中,催化剂粒子的平均粒径具有至少大约0.5mm,在另一个实施例中至少大约0.8mm,以及在另一个实施例中至少大约1.0mm。在一个实施例中,平均粒径的上限位于大约10.0mm,在另一个实施例中大约5.0mm,且在另一个实施例中大约为3.0mm。优选的,催化剂基本由加氢金属,富铈稀土交换分子筛和可选的基质材料组成。更优选的,催化剂基本由一种或多种富铈稀土交换八面沸石,一种或多种第VIII族贵金属,和一种或多种基质材料组成。甚至更优选的,本发明的催化剂基本由一种或多种第VIII族贵金属化合物,一种或多种富铈稀土交换Y型沸石,和一种或多种包括氧化铝的基质组成。催化剂可通过目前工业上已知的工艺来制备,被改进以实现这个发明的特定孔特征。典型的工艺包括连续的步骤:(i)成形,例如挤出固体酸组份,可选择的与基质材料混合之后,形成粒子,(ii)煅烧生成的粒子,以及(iii)引入加氢金属至已煅烧的粒子中,通过例如用加氢金属组份溶液浸渍粒子和/或通过(有竞争性的)离子交换。或者,催化剂可例如通过包括连续步骤的工艺而制备:(i)引入加氢金属到固体酸组份或固体酸组份和基质材料的混合物中,(ii)成形,例如挤出生成的材料以形成粒子,以及(iii)煅烧生成的粒子。关于催化剂制备,在美国2008183025号中描述的流程也可遵循。为了得到本发明的特定孔隙度特征,仔细地进行挤出步骤尤其是有用的。因此,如下所述进行挤出尤其有用的:1)混合基质材料(例如析出的氧化铝粉末),稀土金属交换分子筛(例如沸石),水,硝酸和百分之几的助挤剂(例如甲基纤维素)以形成混合物,2)将这个混合物进料到挤出机中,以及3)根据生成的挤出产品的外观检测,挤出时加入一些额外的水。在实验性的进行这个流程以得到本发明的催化剂时,发现最终挤出混合物中含水量(LOI600)大约为45至55wt%。大约0.05至0.25份的硝酸(相对于氧化铝粉末)被加入。挤出物中的沸石含量大约为65至85wt%,其余为基质以及加氢金属(0.05至0.5wt%Pt),以干燥基计算(600℃,1小时)。本领域普通技术人员目前可以领会到:挤出物获得预期性质(包括机械强度例如侧压强度和堆积强度)需要的具体含水量和加酸取决于使用的分子筛含量和基质材料的特定性质。这通常通过反复试验对起始组份材料测定之后发现。平均粒径长度范围为大约1至大约6mm,粒子直径范围为大约0.5至大约3mm,以及侧压强度范围为大约1至大约10lbs/mm。催化剂尤其适合饱和烃的烷基化。因此本发明进一步有关本发明的催化剂在这些原料的烷基化中的使用。如上所述,这包括饱和烃与烯烃或烯烃前体在本发明的催化剂存在下的反应以产生具有较高分子量的高支链饱和烃。在烷基化工艺中待烷基化的烃是支链饱和烃例如具有4-10个碳原子的异烷烃。实例是异丁烷、异戊烷、异己烷或其混合物。烷化剂是具有2-10个碳原子的烯烃或烯烃混合物。在一个实施例中,烷基化工艺由异丁烷与丁烯的烷基化组成。对于本领域普通技术人员是显而易见的,烷基化工艺可采用任何适合的形式,包括流化床工艺,浆态床工艺和固定床工艺。工艺可在多个床和/或反应器内进行,如果需要,每个均单独加入烷化剂。在这种情况,本发明的工艺可在每个单独的床或反应器内进行。如上所述,在工艺期间水可被加入以提高催化剂的含水量至预期水平。这个水在在烷基化反应期间通过例如烃进料或烷化剂进料来引入。或者,催化剂可在下面描述的可选择的(温和的)再生步骤期间通过使用含水气氛被水化,或在单独的中间水化步骤中通过催化剂接触水被水化。含水量在处理期间(即在烷基化反应和/或再生期间)降低之后,相似的流程可被应用到催化剂的再水化。在根据本发明的工艺中使用的催化剂通过调整含水量被制备。例如,固体酸组成可与基质材料混合以形成载体粒子,接着煅烧粒子。加氢功能物质可以例如通过用加氢金属组份溶液浸渍载体粒子被引入到催化剂组成中。浸渍之后催化剂被煅烧。在一个实施例中,催化剂在温度范围为大约200至大约500℃内在还原性气体例如氢气下被还原。在另一个实施例中,催化剂在温度范围为大约250至大约350℃内被还原。还原可在调节含水量之前,在催化剂中加入水之后进行和/或通过使用还原作为调节含水量的方式。在一个实施例中,在调节含水量之前还原被进行。在另一个实施例中,还原在催化剂在干燥的、非还原气体(例如氮气、氦气、空气等)内干燥之后进行。催化剂的含水量可通过如在PCT/EP2005/000929中描述的各种方法被调节,其通过引用全文的方式被并入。这种方法在如下方法1、2、和3中被举例。方法1涉及通过将催化剂暴露于水提高催化剂含水量。这可通过暴露催化剂于含水气氛例如外界条件的空气来实现。这个方法的实施例包括暴露已还原的催化剂于水直到预期含水量被达到,暴露未还原催化剂于水直到高于预期水平的含水量被达到,接着还原催化剂,从而降低含水量至预期水平,暴露已还原催化剂于水直到高于预期水平的含水量被达到,接着在惰性或还原性气氛中处理催化剂,从而降低含水量至预期水平,以及在氢和含水气氛中还原催化剂。方法2涉及通过还原含水量高于预期水平的未还原的催化剂降低现有催化剂的含水量至预期水平。方法3涉及原位加水,通过用含水量低于预期水平的催化剂开始烷基化工艺且在加工期间向烷基化单元加水,例如通过向烃进料加水,通过在含水气氛下再生催化剂和/或通过暴露已再生的催化剂于含水气氛。两种或多种上述方法的组合也可被应用。适合的工艺条件对于普通技术人员是已知的。优选的,在WO98/23560中描述的烷基化工艺被应用。在本发明中应用的工艺条件总结在下表中:温度范围[℃]压力范围[bar]烃与烷化剂的摩尔比优选的-40–2501–1005:1–5,000:1更优选的20–1505–4050:1–1,000:1最优选的65–9515–30150:1–750:1可选择的,催化剂可用气相的氢气高温再生。这个高温再生可在至少大约150℃的温度下进行,在一个实施例中再生在大约150°至大约600℃下进行,在另一个实施例中在大约200°至大约400℃。这个再生流程的细节,引用的是WO98/23560,尤其是第4页,第12-19行,以全文引用其被并入。在烷基化工艺期间高温再生可定期被应用。如果高温再生的结果是催化剂的含水量被下降到预期水平之下,催化剂在工艺期间可以以上述方式再水化。除了高温再生处理,在烷基化工艺期间温和的再生也可被应用,例如在WO98/23560中描述的,尤其是第9页第13行至第13页第2行,通过引用全文其被并入。在烷基化工艺期间,催化剂通过与含有烃和氢气的进料接触周期地进行再生步骤,在一个实施例中所述再生在催化剂活性周期大约90%或更少下进行,在另一个实施例中大约60%或更少,在另一个实施例中大约20%或更少,以及在另一个实施例中大约10%或更少。在此催化剂的活性周期被定义为从烷化剂开始进料,至与加入到含催化剂的反应器部分的烷化剂相比、20%的烷化剂残留在含催化剂的反应器部分不能被转化的时刻的时间,不包括在分子内的异构化。在一个实施例中,本发明催化剂的制备可包括步骤:a)在温度范围为大约400至大约575℃内煅烧含固体酸粒子;b)引入第VIII族贵金属至已煅烧粒子中以形成含贵金属粒子;以及c)在温度范围为大约350至大约600℃内煅烧含贵金属粒子。或者a)之后,附加的铈通过离子交换和/或浸渍之后干燥和/或煅烧被添加到催化剂中。此后,贵金属被添加。如果铈的引入前后和加氢组分的引入之后煅烧步骤在特定温度窗口被实施,本发明催化剂在烷基化反应中的性能可进一步提高。含固体酸粒子在步骤a)中煅烧的温度范围为大约400至大约575℃内,在另一个实施例中范围为大约450至大约550℃,以及在另一个实施例中范围为大约460至大约500℃。加热速率范围为大约0.1至大约100℃/min,以及在一个实施例中为大约0.5℃至大约50℃/min,以及在另一个实施中为1至大约30℃/min。煅烧在大约0.01至大约10小时内进行,以及在一个实施例中为大约0.1至大约5小时,以及在另一个实施例中为大约0.5至大约2小时。可在空气和/或惰性气体(例如氮气)流内进行。在一个实施例中这个气流是干燥的。在另一个实施例中,含固体酸粒子在被煅烧之前被干燥。这个干燥可在温度为大约110至大约150℃内进行。煅烧可在任何设备中进行,例如固定床反应器,流化床煅烧炉和旋转管煅烧炉。然后第VIII族贵金属在步骤b)中被引入到已煅烧的含固体酸粒子中。在一个实施例中,这可通过使用包括第VIII族贵金属粒子和/或它们的复合物和(可选择的)NH4+离子的溶液对含固体酸粒子的浸渍或竞争的离子交换来进行。在另一个实施例中,第VIII族贵金属为铂、钯和其组合。在另一个实施例中,至少一种第VIII族贵金属是铂。适合的第VIII族贵金属盐包括贵金属的硝酸盐、氯化物、硝酸铵或者它们的络合物(例如NH3络合物)。然后,产生的含贵金属粒子在步骤c)中温度范围为350-600℃内被煅烧。在一个实施例中,粒子在大约400至大约550℃被煅烧,在另一个实施例中,大约450至大约500℃被煅烧。这个温度通过加热粒子以大约0.1至大约100℃/min至达到预期最后值为大约350和大约600℃之间。在一个实施例中,它们以大约0.5至大约50℃/min被加热,在另一个中以大约1至大约30℃/min。煅烧可进行大约0.01至大约10小时,以及在一个实施例中大约0.1至大约5小时,以及在另一个大约0.5至大约2小时。煅烧可在空气和/或惰性气体(例如氮气)流内进行。在一个实施例中这个气体流是干燥的。可选择的,单独干燥步骤被应用于步骤(b)和(c)之间。或者,含贵金属粒子在煅烧步骤期间被干燥。也可选择在采用温度大约200至大约250℃停留15-120分钟。步骤(c)的煅烧之后,生成的催化剂粒子可在温度范围为大约200至大约500℃在还原气体例如氢气下被还原,在一个实施例中从大约250至大约350℃。本发明的催化剂在上述烷基化工艺中的使用导致高烯烃转化率(在进料中的烯烃在反应中被转化的数量),高C5+烷基化物收率(生成的C5+烷基化物的重量除以消耗烯烃的总重量)以及高辛烷值,而不希望得到的C9+副产品数量可被限制因此催化剂的稳定性被提高。关于这些参数的细节,引用WO9823560。下面的实例为了说明而呈现,不是为了限制这个发明的范围。实例测试1:首先,对照例“低Ce”催化剂被制备。根据在US2011/0313227和US8,163,969中描述的流程,沸石用大约4%的“低Ce”稀土(RE)混合物制备。根据在US2011/0313227中描述的流程,使用氧化铝基质大约75%的这个沸石挤出以及用Pt浸渍之后,最终的催化剂含有大约0.1wt%Ce,2.8wt%总RE和2wt%Pt。这个所谓的“低Ce”催化剂根据在US2011/0313227中描述的流程被测试并且与本发明的催化剂对比。然后,本发明具有含较高Ce含量的沸石的催化剂(在沸石上的Ce)被制备。根据在US2011/0313227和US8,163,969中描述的流程,沸石用大约4wt%的“高Ce”稀土混合物制备。根据在US2011/0313227中描述的流程使用氧化铝基质大约75%的这个沸石挤出以及用Pt浸渍之后,最终的催化剂含有大约0.5wt%Ce,3wt%总RE和0.2wt%Pt。这个所谓的“在沸石上的Ce”催化剂根据在US2011/0313227中描述的流程被测试并且与“低Ce”对照例催化剂对比。第三,浸渍附加Ce的本发明催化剂被制备。对照实例的“低Ce”催化剂挤出物样品在浸渍Pt之前,用附加数量的Ce浸渍以提高Ce含量从大约0.1wt%至大约0.5wt%。使用硝酸铈溶液和相对于催化剂的水孔体积大约105wt%饱和度,体积浸渍被进行。使用如在US2011/0313227中描述的与浸渍Pt相似流程,浸渍Ce催化剂被干燥和煅烧。此后,根据如在US2011/0313227中描述的相似流程Pt也被浸渍。最终的催化剂含有大约0.5wt%Ce和大约0.2wt%Pt。这个所谓的“浸渍Ce”催化剂根据在US2011/0313227中描述的流程被测试并且与“低Ce”对照催化剂对比。测试2:根据如在US2011/031322中描述的流程,沸石用大约5wt%的“高Ce”稀土混合物被制备。根据在US2011/0313227中描述的流程使用氧化铝基质挤出大约75%的这个沸石以及用Pt浸渍之后,最终的催化剂含有大约1wt%Ce,3.8wt%总RE和0.2wt%Pt。第二个样本催化剂以相似的方式被制备且制备具有2wt%Ce,3.8wt%总RE和0.2wt%Pt。最后,第三样本催化剂使用相似方法被制备且制备仅具有4.8wt%的Ce。测试1和测试2的已测试烷基化催化剂具有下面的组成和性能:上述沸石从大约60至大约75%,氧化铝从大约25%至大约40%,Pt从大约0.2wt%至大约0.5%,平均粒径长度范围从大约1至大约6mm,平均长度/直径比范围从大约1至大约12,粒径范围从大约0.5至大约3mm,以及侧压强度范围从大约1至大约10lbs/mm。常规测试流程:如在WO9823560中描述的固定床循环反应器,在此其以全文引用被并入,具有直径2cm,被填充体积/体积为1:1的38.6克的催化剂挤出物(干燥基,LOI600)和碳化硅颗粒(60目)的混合物。在反应器管中间直径6mm的热电偶被配置。反应器用干燥氮气冲扫30分钟(21Nl/hour)。接着,系统在高压下进行泄露测试,之后设定压力至21bar以及氮气流至21Nl/hour。然后反应器温度以1℃/min的速率被提高至275℃,在275℃氮被干燥氢取代且275℃催化剂被还原。或者,在同样催化剂样品在使用期间高温再生运行的情况下,用氢气排出和冲扫以除去烃之后且同时保持烷基化反应温度,氢气流被设置为21Nl/hour并且反应器温度以1℃/min的速率被提高至275℃,在275℃催化剂被还原。2小时之后,反应器温度被降低至大约75℃的反应温度。在冷却期间水被加入到氢气流以得到含水量为大约2-4wt%的催化剂(被定义为在600℃加热2小时之后催化剂的水分损失)。到达反应温度时氢气流停止。含大约4wt%的烷基化物的异丁烷(被加入以加速失活速率,加入的烷基化物的组成与在描述的条件下的工艺中生产的烷基化物相似)以及大约1mol%的溶解氢以速率为4.0kg/hour被提供到反应器。大约95-98%的异丁烷/烷基化物混合物被进料到反应器。大约2-5%被排出用于分析。这样数量的异丁烷/烷基化物混合物被提供到反应器以确保在系统中的恒定量液体。当系统已经稳定,停止氢的添加且这种数量的顺-2-丁烯被加入到它中使得顺-2-丁烯-质量空速(WHSV)为0.16。系统中的液体总流速被保持在大约4.0kg/h。在反应器内异丁烯与顺-2-丁烯的重量比为大约600–700。反应器内的压力总计为大约21bar。在测试期间,通过控制用于分析的排出流烃循环流的总烷基化物浓度(从加入的和产生的烷基化物中)被保持在大约8-9wt%。每次1小时反应之后,催化剂被再生:通过用异丁烷/烷基化物混合物洗涤5分钟,接着通过与在异丁烷/烷基化物混合物中的1mole%的H2溶液接触进行50分钟的再生,以及用异丁烷/烷基化物混合物再洗涤5分钟(洗涤和再生总时间1小时)。这个洗涤步骤之后,烷基化被再次开始。洗涤步骤,再生步骤以及反应步骤期间的温度相同。工艺如上所述被进行且催化性能随时间的变化被测试。性能以每个反应器通过的烯烃转化率进行表征。每个反应器通过的烯烃转化率是在催化床的入口和出口之间被转化烯烃的重量百分率(百分比),不算异构化烯烃分子。测试1中得到的各种催化剂结果在图1和图2中呈现。图1示出随进行时间变化的烯烃转化率。图2示出随进行时间变化的C9+复合物(重质)。测试2中得到的各种催化剂结果在图3和图4中呈现。从这些结果,可以得出结论:比0.5wt%Ce更高的样品给出相似的结果。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1