分析甲磺酸乐伐替尼及其制剂杂质的HPLC方法及杂质作参比标准的用途与流程
本发明属于药物合成领域。具体地,本发明涉及乐伐替尼(4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺)制备过程中出现的杂质及对含有乐伐替尼制剂进行分析的方法。
背景技术:
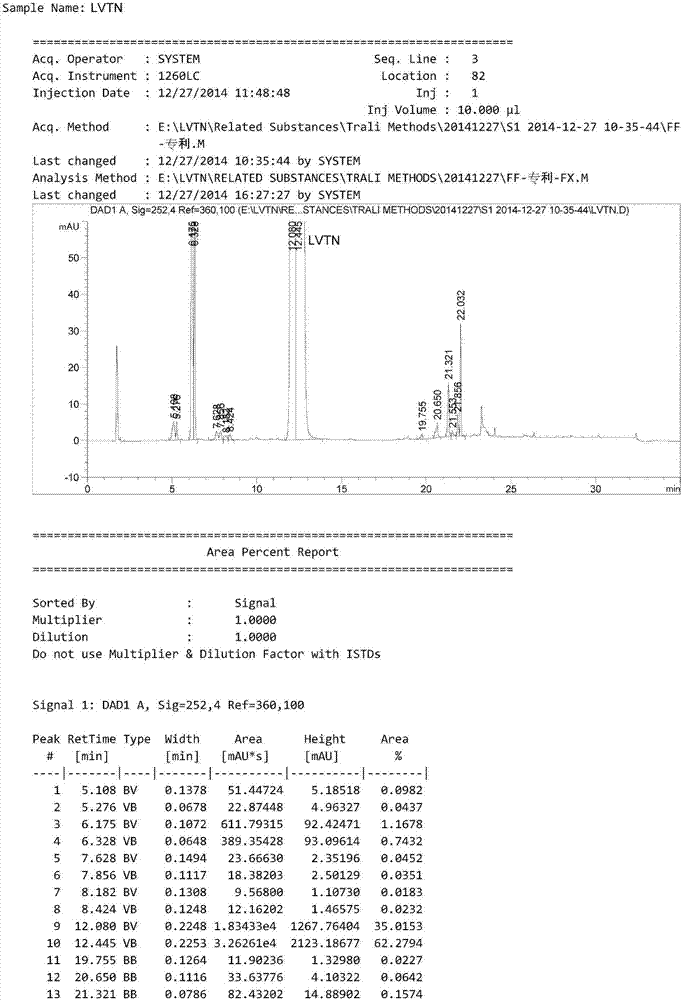
:lenvatinib由卫材公司研制并开发的,目前尚无标准的中文译名,因此本申请人在此将其音译为“乐伐替尼”。该药已于2015年2月获美国fda批准,用于有局部地复发或转移,进展性,放射性碘难治性分化型甲状腺癌患者的治疗,商品名lenvima。2015年3月,乐伐替尼被日本厚生省批准上市,成为治疗不可切除性甲状腺癌(包括分化型甲状腺癌、甲状腺髓样癌、未分化型甲状腺癌)的首个分子靶向治疗药物;2015年5月,被欧洲食品药品监督管理局批准上市。lenvima是一种口服给药的分子靶向制剂。目前尚无标准的中文译名,因此本申请人在此将其音译为“甲磺酸乐伐替尼”。乐伐替尼的化学名称为:4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺,其甲磺酸盐的结构式为:专利cn200480036184.1中公开了对乐伐替尼或乐伐替尼不同盐的化学纯度进行分析的hplc方法。按照该专利的试验例3方法对乐伐替尼粗品(本专利实施例2中制备的未经乙醇重结晶的)进行分析,所使用的高效液相色谱仪(hplc)为agilent1260,pda检测器(购自安捷伦公司),结果如图1所示,其中图1中的最高峰为乐伐替尼的峰。由图1可知,该专利的分析方法存在如下弊端:(1)乐伐替尼粗品中含有的杂质(该方法下保留时间约为:5.063min、5.180min和5.273min)相互之间不能实现基线分离,无法进行准确定量;杂质之间的分离度不符合ichq3a中对于有关物质方法学验证项下对专属性的要求,即杂质之间的分离度小于1.5;(2)该方法明确所用溶剂为水-甲醇(v/v,3∶1),供试品溶液的浓度为0.1mg/ml,这样低的供试品溶液浓度,势必造成一些指定波长下紫外吸收弱的杂质检测不到,造成杂质低估。当将该条件下供试品溶液的浓度提高至0.5mg/ml时,采用专利中的溶剂不能完全溶解,加二甲基亚砜(dmso)助溶后,在该方法下进样分析,多个谱峰为分叉峰,具体见图2。(3)该方法杂质的计算方法为面积归一化法,由于各杂质结构不同,在拟定的检测波长下,紫外吸收情况是不同的,因此,面积归一化法并不能准确测定出各杂质在上市药品中的真实含量。(4)并没有对甲磺酸乐伐替尼中的特定杂质进行定性分析。综上所述,现有技术的检测结果并不能真实反映药物的质量。技术实现要素:本发明提供了新的、可替换的方法,用于乐伐替尼或含乐伐替尼的制剂中有关物质的定性、定量分析,并用这些杂质作为参比标准或对照品来进行杂质的定量和定性分析。本发明的另一个目的是提供参比标准或对照品,用于检测在制备乐伐替尼的过程中或制剂工艺及贮藏过程中形成的称为a~i及lvtn-1杂质。本发明提供的用于分析乐伐替尼或乐伐替尼的制剂中有关物质的hplc方法。发明人利用9种制备和鉴定结构的杂质(称化合物a~i及lvtn-1)用于乐伐替尼或含有乐伐替尼的药物制剂有关物质分析方法的参比标准或对照品。其中包括现有技术中已经公开的六种杂质及结构和现有技术中尚未披露的三种杂质及结构。因此,本发明的分析方法用于分析化合物a,其具有化学名:4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺),并具有以下结构:第二个方面提供化合物b,其具有化学名:4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺,并具有以下结构:第三个方面提供化合物c,其具有化学名:4-乙氧基-7-甲氧基喹啉-6-甲酰胺,并具有以下结构:第四个方面提供化合物d,其具有化学名:4-羟基-7-甲氧基喹啉-6-羧酸酰胺,并具有以下结构:第五个方面提供化合物e,其具有化学名:1-(2-氯-4-羟基苯基)-3-环丙氨基脲,并具有以下结构:第六个方面提供化合物f,其具有化学名:4-(3-氯-4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸,并具有以下结构:第七个方面提供化合物g,其具有化学名:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸甲酯,并具有以下结构:第八个方面提供化合物h,其具有化学名:4-(4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺,并具有以下结构:第九个方面提供化合物i,其具有化学名:4-(3-氯-4-(3-甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺,并具有以下结构:第十个方面提供甲磺酸乐伐替尼制备中间体-1(lvtn-1),其具有化学名:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺,并具有以下结构。化合物a~i和中间体-1(lvtn-1)为甲磺酸乐伐替尼合成过程中的中间体或副产物或甲磺酸乐伐替尼及含有甲磺酸乐伐替尼制剂组合物贮存过程中的降解产物,适用于作参比标准或对照品。在一个特别优选的实施方式中,根据本发明的化合物a~i和中间体-1(lvtn-1)为单体化合物,最优选的是纯的形式,优选纯度高于约95%、优选纯度高于约98%、最优选纯度高于约99%,优选通过hplc测量。根据本发明的第十个方面提供一种检测甲磺酸乐伐替尼或含有甲磺酸乐伐替尼的药物剂型的样品纯度的方法,该方法包括测定样品中根据本发明的化合物a~i和中间体-1(lvtn-1)中的一种或多种的存在。在本发明的方法中,所述化合物用作参比标准或对照品。根据本发明的第十一个方面,提供一种用于表征化合物a~i和中间体-1(lvtn-1)的方法,该方法利用hplc方法来分析乐伐替尼中的所述杂质a~i和中间体-1(lvtn-1)。优选该hplc方法为与lc-ms相容的hplc方法。另一方面提供一种用于检测乐伐替尼或含有乐伐替尼的制剂样品纯度的色谱方法,所述方法包括:通过利用根据本发明的参比标准或对照品,来测定样品中化合物a~i和中间体-1(lvtn-1)中的一种或多种的存在。化合物a~i和中间体-1(lvtn-1)为甲磺酸乐伐替尼合成过程中的工艺杂质、副产物或降解杂质,适用于作为参比标准或对照品,用于该产品的质量控制。以上杂质的产生途径如下:杂质a:本杂质为市售胶囊剂中杂质,应该为原料药甲磺酸乐伐替尼引入的工艺杂质,即甲磺酸乐伐替尼制备工艺第二步生成的杂质。杂质b:在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法第三步与环丙胺的反应中,由于采用的溶剂为n,n-二甲基甲酰胺,其中可能含有少量的二甲胺生成的杂质b。杂质c:中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法第4步成盐反应中,酸性条件下,中间体-3(lvtn-3,即乐伐替尼游离碱)被溶剂乙醇进攻生成杂质c。杂质d和杂质e:在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法第四步成盐的反应中,在酸性条件下,乐伐替尼游离碱(lvtn-3)发生分解,生成杂质d和杂质e,二者即为工艺杂质,也是降解杂质。杂质f(lvtn-zz-10):甲磺酸乐伐替尼中含有酰胺基团,在酸或碱的条件下可以水解为羧酸,为降解杂质。杂质g:甲磺酸乐伐替尼在酸、碱、湿热条件下降解产生降解杂质g。酸降解机理:杂质h:在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法中,起始物料2脱卤杂质后续反应生成。杂质i:在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法中第三步反应中,溶剂n-甲基吡咯烷酮与环丙胺中可能含有少量甲胺,与环丙胺形成竞争,参与取代反应。产生机理如下:中间体-1(lvtn-1):在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法中第一步中间体,也是甲磺酸乐伐替尼贮藏或后续工艺步骤中的降解杂质。其产生机理如下:(1)在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法中的第二步,lvtn-1过量,剩余。(2)在中国专利申请号cn101024627报道的甲磺酸乐伐替尼的合成方法的第三步成盐时,lvtn-3遇酸和温度作用降解。酸降解机理:杂质d、e、f、g、lvtn-1为甲磺酸乐伐替尼或包含甲磺酸乐伐替尼的药物制剂中的主要降解杂质。在一个特别优选的实施方式中,根据本发明的化合物a~i和lvtn-1为单体化合物,最优选的是纯的形式,优选纯度高于约95%、优选纯度高于约98%、最优选纯度高于约99%,优选通过hplc测量。根据本发明的第十个方面提供一种检测乐伐替尼或含有乐伐替尼的药物剂型的样品纯度的方法,该方法包括测定样品中含有本发明的化合物a~i和lvtn-1中的一种或多种。在本发明的方法中,所述化合物用作杂质的参比标准或对照品。根据本发明的第十个方面,提供一种用于表征化合物a~i和lvtn-1的方法,该方法利用hplc方法来分析乐伐替尼中的所述杂质a~i和lvtn-1。优选该hplc方法为与lc-ms相容的hplc方法。因此,提供了根据本发明的化合物a~i和lvtn-1(一种或多种)在检测乐伐替尼或含有乐伐替尼的药物制剂的样品纯度中作为参比标准或对照品的用途。另一方面,本发明还提供一种用于检测乐伐替尼样品纯度的色谱方法,所述方法包括:通过利用根据本发明的参比标准或对照品,来测定样品中化合物a~i和lvtn-1中的一种或多种的存在。又一个方面,提供一种通过测定含有乐伐替尼的样品中化合物a~i和lvtn-1中任何一种或多种的存在来检测甲磺酸乐伐替尼样品的纯度的色谱方法,所述方法包括:(1)将乐伐替尼或含有乐伐替尼的制剂样品溶解在溶剂中以制备样品溶液;(2)将化合物a~i和lvtn-1中任何一种或多种的样品溶解在溶剂中以制备参比标准溶液或对照品溶液;(3)对样品溶液和参比标准溶液实施色谱技术;以及(4)通过参照该参比标准溶液中存在的已知化合物a-i和lvtn-1(一种或多种),测定样品中化合物a~i中的任何一种或多种的存在。在一种实施方式中,该色谱方法为液相色谱法,如hplc、uplc、lc-ms;优选该色谱法为hplc法,优选梯度hplc法。本发明优选使用的固定相为反相。合适的固定相包括十八烷基硅烷键合硅胶或辛基硅烷键合硅胶。在本发明的优选实施方式中,提供一种梯度hplc方法,其中,流动相包括含缓冲溶液(a)和有机溶剂(b)的组合。优选缓冲溶液(a)为含水缓冲液,优选为醋酸盐、甲酸盐、磷酸盐、三氟醋酸、甲酸或它们的混合物的水溶液。更优选的缓冲溶液(a)为醋酸盐,最优选为醋酸铵和醋酸或醋酸铵的水溶液用醋酸调节过ph的水溶液。在特别优选的实施方式中醋酸铵存在的浓度约为0.001m至1.0m,优选0.02m至0.08m,更优选0.02m至0.05m,最优选0.03m。优选有机溶剂(b)为极性质子溶剂,如甲醇、乙醇或异丙醇;或偶极非质子溶剂,如乙腈。优选有机溶剂(b)选自包括甲醇、乙腈、乙醇、异丙醇或它们的混合物的组中,优选的为甲醇和乙腈的组合,其中以流动相a和b的总百分比计,甲醇的百分比为25±5%。本发明特别优选的流动相包含醋酸铵和醋酸的水溶液(a)和乙腈(b)的组合。进一步提供根据本发明的梯度hplc方法,其中流动相包含如下的梯度设计:时间(分钟)流动相a(%)流动相b(%)0901087525304060355953759537.019010459010进一步优选的实施方式中,hplc方法中的缓冲溶液(a)醋酸铵和醋酸的水溶液的ph为约3.5~6.5,优选为约4.0~6.5,4.0~6.0,4.0~5.5,最优选为4.5~5.5。在其它实施方式中,该hplc分析方法在约20~40℃的温度下进行,优选为20~35℃,最优选为25~35℃。在其它实施方式中,该分析方法在约0.3~1.0ml/min的流速下进行,优选为0.3~0.9ml/min,0.3~0.8ml/min,0.3~0.7ml/min,0.3~0.6ml/min,最优选为0.3~0.5ml/min。根据本发明的hplc方法以单次操作有效地检测并定量包括那些选自以下化合物中的化合物的所有杂质:化合物a:4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺)。化合物b:4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺。化合物c:4-乙氧基-7-甲氧基喹啉-6-甲酰胺。化合物d:4-羟基-7-甲氧基喹啉-6-羧酸酰胺。化合物e:1-(2-氯-4-羟基苯基)-3-环丙氨基脲。化合物f:4-(3-氯-4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸。化合物g:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸甲酯。化合物h:4-(4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺。化合物i:4-(3-氯-4-(3-甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺。lvtn-1:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺.本发明可用于分析作为活性药物成分(api)的乐伐替尼和/或药物组合物中的乐伐替尼。本发明能够分析的药物组合物包括固体或液体组合物,并可选地包括一种或多种药学上可接受的赋形剂。固体形式的组合物包括粉剂、片剂、胶囊剂、丸剂以及可分散的颗粒剂等。液体组合物包括溶液或悬浮液,其可通过口服、注射或滴注途径给药。说明书与权利要求书通篇所使用的术语“乐伐替尼”是指4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺或其溶剂化物(如水合物)和/或其盐,包括甲磺酸盐。说明书通篇所使用的术语“杂质”或“有关物质”可意指在制造api或药物组合物中形成的杂质,以及/或者api降解形成的杂质或在储存中的药物组合物或制剂中形成的杂质。如上所述,现有技术中所报道的hplc方法,不适合用于分析乐伐替尼及含有乐伐替尼的制剂或药物组合物。然而,本发明的方法解决了这一问题,并且以单次操作有效地检测并定量、定性分析这种特定合成过程或制剂制备中形成的所有杂质。本发明的优势在于:披露了乐伐替尼和乐伐替尼制剂中的杂质的具体结构,并运用梯度hplc法同时将极性杂质和非极性杂质洗脱出来,并进行定性、定量分析。本发明特别适合于测定和定量样品中化合物a~i和lvtn-1中的一种或多种的存在。除非另有描述,否则本文中的术语“杂质”和“化合物”在与化合物a~i和lvtn-1相关的范围下,可以互换使用。本发明的优势还在于,这种方法对于乐伐替尼及含乐伐替尼的制剂中的有关物质的分析具有专属性强,准确度、精密度高、耐用性好等特点。此外,本发明具有高度灵敏度,且允许检测和定量乐伐替尼api或药物组合物中水平量显著低于ich指导原则中规定的可接受限值的有关物质。此外,本发明的方法可用于检测和定量乐伐替尼样品或药物组合物储存过程中形成的所有降解杂质。通过按照ichq1a指南进行强制降解研究来确定该方法,并按照ichq2a指南进行验证,该验证覆盖以下项目:专属性、线性与范围、精密度、准确度、检测限、定量限、耐用性及系统适应性。本发明人开发出的新型梯度hplc方法已定性测定九种杂质a~i和lvtn-1。所述方法能够一次性分析按照本实施例制备的乐伐替尼制备工艺中以及市售乐伐替尼及储存过程中产生的降解杂质等极性差异较大的杂质,因此,本发明人认为梯度设计最为合适。在实施本发明中,本发明的发明人发现含有十八烷基硅烷键合硅胶或辛基硅烷键合硅胶的固定相最为有利。特别优选的固定相含有watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm。本发明方法优选包括梯度设计,使得流动相a和b的相对浓度在10~60分钟时间典型地变化为100%a:0%b至0%a:100%b的梯度。优选地,经过30至55分钟时间,梯度为95%a:5%b至5%a:95%b,更优选地,经过20至50分钟时间,梯度为90%a:10%b至15%a:85%b,最优选地,经过约30分钟,梯度为90%a:10%b至15%a:85%b或85%a:15%b至20%a:80%b或80%a:20%b至30a%:70%b或70%a:30%b至40%a至60%b。这种梯度法的优势在于,能够将乐伐替尼api或乐伐替尼药物组合物中各种不同极性或极性非常相近的杂质全部分离开,便于准确的定性和定量。使用的流动相优先选自一种或多种缓冲溶液(a)和一种或多种有机溶剂(b)的组合。缓冲溶液(a)优先选自包括磷酸盐、醋酸盐、甲酸盐、三氟醋酸、甲酸或醋酸它们的混合物的水溶液组合。缓冲溶液(a)的浓度可以为0.001m至1.0m,优选浓度为0.02m至0.08m,更优选浓度为0.02m至0.05m,最优选0.03m。特别优选的流动相包含醋酸铵和醋酸的水溶液,或醋酸铵溶液用醋酸调节ph的水溶液(a)和乙腈(b)的组合。在根据本发明的特别优选的实施方式中,进一步提供一种梯度hplc的方法,其中流动相包括如下的梯度设计:本发明还提供的特别优选的hplc梯度方法,其中,流动相包含醋酸铵和/或醋酸作为缓冲溶液(a)。在另外特别优选的实施方式中,流动相包含乙腈作为有机溶剂(b),和/或乙腈-甲醇混合溶剂作为有机溶剂(b)。发明人发现当流动相包含醋酸铵和/或醋酸(a)和乙腈(b)时梯度设计尤其有效。缓冲溶液(a)可含有一种或几种附加溶剂,这些附加溶剂可以是甲醇、乙醇、异丙醇、乙腈或它们的混合物作为有机溶剂。缓冲溶液(a)中的附加溶剂可以是或不是与有机溶剂(b)相同的溶剂。缓冲溶液(a)中的附加溶剂优选为乙腈。缓冲溶液(a)的ph为约3.5~6.5,优选为约4.0~6.5,4.0~6.0,4.0~5.5,最优选为4.5~5.5。本发明的hplc方法在约20~40℃的温度下进行,优选为20~35℃,最优选为25~35℃。该分析方法在约0.3~1.0ml/min的流速下进行,优选为0.3~0.9ml/min,0.3~0.8ml/min,0.3~0.7ml/min,0.3~0.6ml/min,最优选为0.3~0.5ml/min。本发明的另一方面提供一种参比标准溶液。该溶液包含溶解于合适溶剂(如乙腈)中的一种或多种化合物a~i和lvtn-1。所述参比标准溶液可用于测定在利用根据本发明的色谱技术进行分析的样品中作为杂质的任何化合物a~i和lvtn-1的存在。根据本发明的另一方面,提供一种参比标准溶液,其中,已知量的一种或多种化合物a~i和lvtn-1溶解于合适的溶剂(如乙腈或二甲基亚砜或二者的混合物)中。所述参比标准溶液可用于测定在利用根据本发明的色谱技术进行分析的样品中作为杂质的任何化合物a~i和lvtn-1的定性和定量。所述分析的方法对于技术人员的重要和方便是显而易见的。本发明人已广泛验证本发明的方法,验证结果表明本方法的专属性强、准确度、精密度和灵敏度高,耐用性好。附图说明:图1为按照cn200480036184.1试验例3公开的方法对乐伐替尼进行hplc分析图谱;图2:为按照cn200480036184.1试验例3公开的方法,加大乐伐替尼供试品溶液浓度后进行hplc分析图谱;图3:中间体-1(lvtn-1):4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的1h-nmr;图4:中间体-1(lvtn-1):4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的高分辨质谱(esi-hrms);图5:甲磺酸乐伐替尼:4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺甲磺酸盐的1h-nmr;图6:4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺甲磺酸盐的高分辨质谱;图7:杂质a:4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺)的1h-nmr;图8:杂质a:4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺)的高分辨质谱;图9:杂质b:4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的1h-nmr;图10:杂质b:4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的高分辨质谱;图11:杂质c:4-乙氧基-7-甲氧基喹啉-6-甲酰胺的1h-nmr;图12:杂质c:4-乙氧基-7-甲氧基喹啉-6-甲酰胺的高分辨质谱;图13:杂质d:4-羟基-7-甲氧基喹啉-6-羧酸酰胺的1h-nmr;图14:杂质d:4-羟基-7-甲氧基喹啉-6-羧酸酰胺的高分辨质谱;图15:杂质e:1-(2-氯-4-羟基苯基)-3-环丙氨基脲的1h-nmr;图16:杂质e:1-(2-氯-4-羟基苯基)-3-环丙氨基脲的高分辨质谱;图17:杂质f:4-(3-氯-4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸的1h-nmr;图18:杂质f:4-(3-氯-4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸的高分辨质谱;图19:杂质g:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸甲酯的1h-nmr;图20:杂质g:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸甲酯的高分辨质谱;图21:杂质h:4-(4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的1h-nmr;图22:杂质h:4-(4-(3-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的高分辨质谱;图23:杂质i:4-(3-氯-4-(3-甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的1h-nmr。图24:杂质i:4-(3-氯-4-(3-甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的高分辨质谱;图25:供试品加标混合溶液的hplc谱图;图26:本发明专利实施例3制备的甲磺酸乐伐替尼供试品溶液的hplc谱图;图27:自制的甲磺酸乐伐替尼胶囊-10mg供试品溶液的hplc谱图;图28:市售甲磺酸乐伐替尼胶囊-10mg供试品溶液的hplc谱图;具体实施方式虽然本发明已对其具体实施方式进行描述,然而某些修改和等同物对于本领域技术人员是显而易见的,并且旨在包括在本发明的范围之内。通过以下实施例对本发明进行阐述,以下实施例不以任何方式限制本发明。实施例1:4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺(lvtn-1)的合成。氮气保护下,向500ml的三口瓶中,加入200ml二甲基亚砜,开启搅拌,依次加入20.00g4-氯-7-甲氧基喹啉-6-羧酸酰胺(sm1),18.20g4-氨基-3-氯苯酚(sm2)及14.22g叔丁醇钾。加料完毕,升温至65℃,保温搅拌19小时。将上述反应体系倾倒入纯净水中,有大量固体析出,过滤,滤饼用纯净水淋洗,在60℃条件下,鼓风干燥1~2小时,得到棕色固体25.07g4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺(lvtn-1),收率:86.32%。中间体-1的1h-nmr和高分辨质谱分别参见图3和图4。1h-nmr(400mz,dmso)δ:8.685(s,1h),8.653(d,j=3.6hz,1h),7.754-7.870(br,s,1h),7.510(s,1h),7.263(d,j=2.0hz,1h),7.023(dd,j=2.0hz,j=6.0hz1h),6.931(d,j=6.0hz,1h),6.472(d,j=3.2hz,1h),5.479(s,2h),4.042(s,3h)。esi-hrms谱图显示分子离子峰m/z=344.08046[m+h]+,所对应的分子量与提供的结构式理论计算值(344.07237)相符。绝对误差为2.38ppm,在高分辨质谱误差范围之内。实施例2:4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺(lvtn-3)乐伐替尼的合成向500ml三口瓶中加入250mln-甲基吡咯烷酮,开启搅拌,再依次加入11.50g吡啶和25.00g4-(4-氨基-3-氯苯氧基)-7-甲氧基喹啉-6-羧酸酰胺(lvtn-1)。冰水浴下,向体系中滴加4.94g氯甲酸苯酯,滴加完毕后,将体系升温至室温。向反应体系中滴加纯净水,滴加时间1~2小时,过滤,淋洗,于60℃条件下鼓风干燥2小时,得到33.74g(4-((6-羧酸甲酰胺基-7-甲氧基喹啉-4-基)氧)-2-氯苯基)氨基甲酸苯酯(lvtn-2)棕色粉末。收率:81.26%。向1000ml三口瓶中加入300mln-甲基吡咯烷酮,开启搅拌,再加入30.00g(4-((6-羧酸甲酰胺基-7-甲氧基喹啉-4-基)氧)-2-氯苯基)氨基甲酸苯酯(lvtn-2)。室温下,向体系中滴加4.43g环丙胺,滴加时间1小时。滴加完毕后,继续保温搅拌过夜。向反应体系中加入300ml纯净水,搅拌至有大量固体析出(约1~2小时),继续搅拌1小时,过滤,淋洗,于60℃条件下鼓风干燥12小时,得到27.50g粗品,经乙醇重结晶得到21.08g类白色固体4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺(lvtn-3),收率:76.35%实施例3:甲磺酸乐伐替尼的制备,4-(3-氯-4-(n’-环丙基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺甲磺酸盐的制备以及甲磺酸乐伐替尼胶囊的制备氮气保护下,向250ml三口瓶中依次加入100ml醋酸和2.70g甲磺酸,控温40℃条件下,再向体系中加入10.00glvtn-3,搅拌1~2小时,过滤,收集母液,将母液转移500ml三口瓶中,氮气保护下,控温40℃,向体系中滴加170ml正丙醇,控制滴加时间1~2小时。过滤,滤饼用70ml正丙醇、淋洗,得到甲磺酸乐伐替尼的醋酸合物。氮气保护下,向250ml三口瓶中加入100ml乙醇,开启搅拌,控温40℃条件下,加入甲磺酸乐伐替尼醋酸合物,继续搅拌36小时,过滤,滤饼用乙醇淋洗,于60℃条件下,鼓风干燥12小时,得到甲磺酸乐伐替尼成品10.23g,为类白色粉末,收率:83.49%。1h-nmr(400mz,cdcl3)δ:8.975(d,j=6.8hz,1h),8.715(s,1h),8.354(d,j=9.2hz,1h),8.079(s,1h),7.971(br,s,1h),7.909(br,s,1h),7.699(s,1h),7.638(d,j=2.4hz,1h),7.359(dd,j=2.4hz,j=9.2hz,1h),7.274(s,1h),6.963(d,j=6.8hz,1h),4.081(s,3h),2.572~2.578(m,1h),2.401(s,1h),0.651~0.665(m,2h),0.429(m,2h)。esi-hrms谱图显示分子离子峰m/z=427.11803[m+h]+,所对应的分子量与提供的结构式理论计算值(427.10948)相符。绝对误差为2.98ppm,在高分辨质谱误差范围之内。甲磺酸乐伐替尼1h-nmr谱图和高分辨质谱(esi-hrms)图见图5~6。称取如下处方量的甲磺酸乐伐替尼、d-甘露醇、沉降碳酸钙、羟丙基纤维素、低取代羟丙基纤维素和微晶纤维素ph101,置高速搅拌混合制粒机中混合,加纯化水适量制软材,过18目筛制粒,流化床干燥至水分含量低于2.0%,置干颗粒快速整粒机整粒,筛网目数14目,整粒后颗粒加入微晶纤维素ph102、滑石粉混合,填充4号羟丙甲基纤维素胶囊,由此制备得到甲磺酸乐伐替尼胶囊。处方组成用量(mg/粒)甲磺酸乐伐替尼12.5沉淀碳酸钙33d-甘露醇8.5微晶纤维素ph10110低取代羟丙基纤维素25羟丙基纤维素-l3微晶纤维素ph1025滑石粉3实施例4:杂质a的合成杂质a,4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺)的合成向100ml的三口瓶中加入20mln-甲基吡咯烷酮,搅拌下依次加入1.00glvtn-1,0.92g吡啶,氮气保护,降温至0~10℃,开始滴加1.46g氯甲酸苯酯,滴毕,继续搅拌20~30分钟,升温至60℃,搅拌过夜;取样tlc检测(甲醇∶二氯甲烷=1∶10)原料消失。加水20ml,有大量固体析出,继续搅拌30分钟。过滤,抽干,40℃鼓风干燥30分钟,得到固体粗品,将得到粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶50,收集洗脱液共160ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,得淡黄色固体105mg,收率:5.05%,即得到杂质a:4,4′-(((羰基双(脲二基))双(3-氯-4,1-苯基))双(氧基))双(7-甲氧基喹啉-6-甲酰胺)。杂质a的结构确证如下:表1杂质a的1h-nmr和13c-nmr测试数据(dmso-d6)和归属杂质a的esi-hrms谱图显示分子离子峰[m+h]+的质荷比为713.13146,所对应的分子量与提供的结构式理论计算值(713.12400)相符。绝对误差为0.26ppm,在规定的误差范围之内。杂质a的1hnmr和esi-hrms谱图分别见附图7-8。实施例5:杂质b的合成杂质b,即4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺的合成。向50ml的三口瓶中加入463.87mg甲磺酸乐伐替尼中间体lvtn-2和9mln-甲基吡咯烷酮。控制反应温度在0~10℃,向反应体系中加入99.18mg二甲胺,继续搅拌30分钟,tlc监测(二氯甲烷∶甲醇=10∶1),反应结束。向反应体系中加入80%丙酮/水(v/v)混合溶剂18ml,有大量固体析出,搅拌30分钟后,过滤,60℃鼓风干燥,得到杂质b粗品。将该粗品进行柱层析纯化,展开剂及比例为:甲醇∶二氯甲烷=1∶20(v/v),收集洗脱液共150ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,至无馏分蒸出,得淡粉色固体153.04mg,收率:36.89%,即得到杂质b:4-(3-氯-4-(3,3-二甲基脲基)苯氧基)-7-甲氧基喹啉-6-羧酸酰胺。1h-nmr(400mz,cdcl3)δ:8.709(d,j=3.2hz,1h),8.678(s,1h),7.981(s,2h),7.874(s,1h),7.724(d,j=6.0hz,1h),7.544(s,1h),7.274(dd,j=2.0hz,j=6.0hz,1h),7.266(d,j=2.0hz,1h),6.559(d,j=3.2hz,1h),4.051(s,3h),2.978(s,6h)。esi-hrms谱图显示分子离子峰m/z=415.11742[m+h]+,所对应的分子量与提供的结构式理论计算值(414.84226)相符。绝对误差为1.60ppm,在高分辨质谱误差范围之内。杂质b的1hnmr和esi-hrms谱图分别见附图9-10。实施例6:杂质c的合成杂质c,即4-乙氧基-7-甲氧基喹啉-6-甲酰胺的合成向100ml的三口瓶中加入2.00g甲磺酸乐伐替尼,50ml乙醇。回流搅拌72小时,tlc监测(甲醇∶二氯甲烷=1∶10,有新点产生)。减压蒸除溶剂,将得到粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶20,收集洗脱液共60ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,得淡粉色固体122mg,收率:6.10%,即得到杂质c:4-乙氧基-7-甲氧基喹啉-6-甲酰胺。杂质c的结构确证见下表:表2杂质c的1h-nmr和13c-nmr测试数据(dmso-d6)和归属杂质c的esi-hrms谱图显示分子离子峰[m+h]+的质荷比为247.10872,[2m+na+h]+的质荷比为515.19080,所对应的分子量与提供的结构式理论计算值(247.10044)相符。绝对误差为4.08ppm,在规定的误差范围之内。杂质c的1hnmr和esi-hrms谱图分别见附图11-12。实施例7:杂质d的合成向250ml三口瓶中加入10.00g的lvtn-3,50ml乙腈,控温80℃,搅拌,加6%双氧水3ml,继续加热15分钟,淬灭,浓缩。将该粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶10,收集洗脱液共720ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,减压蒸馏,蒸除溶剂至无馏分蒸出,得化合物d1.12g,化合物d为黄色粉末,hplc纯度:99.19%。杂质d的1hnmr和esi-hrms谱图分别见附图13-14。实施例8:杂质e外购于南京奇可医药化工有限公司,hplc纯度为99.06%。杂质e的1hnmr和esi-hrms谱图分别见附图15-16。实施例9:杂质f的合成向100ml三口瓶中加入lvtn-3约2.00g,加乙腈20ml、2mol/l盐酸溶液,超声使分散均匀,于80℃水浴加热30分钟,浓缩。将该粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶10,收集洗脱液共620ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,减压蒸馏,蒸除溶剂至无馏分蒸出,得淡黄色粉末0.66g,hplc纯度:98.15%。杂质f的1hnmr和esi-hrms谱图分别见附图17-18。实施例10:杂质g的合成向250ml的三口瓶中加入5.00glvtn-2,50mln-甲基吡咯烷酮。控温0~10℃,加入7.56g氨水,继续搅拌30分钟,tlc监测(二氯甲烷∶甲醇=10∶1),反应结束。加入33%丙酮/水75ml,有大量固体产生,搅拌30分钟后,过滤,60℃鼓风干燥,得到化合物g粗品。将该粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶20,收集洗脱液共630ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,得淡黄色固体0.86g,收率:20.63%,即得到化合物g,hplc纯度:99.60%。杂质g的1hnmr和esi-hrms谱图分别见附图19-20。实施例11:杂质h的合成(1)向250ml的三口瓶中加入15ml二甲基亚砜,搅拌下,依次加入3.00g4-氯-7-甲氧基喹啉-6-羧酸酰胺(sm1)和1.66g4-氨基苯酚(sm2-zz-02)。控温20~30℃,加入1.71g叔丁醇钾,继续搅拌14小时,并取样做tlc监控(二氯甲烷∶甲醇=10∶1),反应结束。加入30ml纯净水,有大量固体析出,过滤,得到粗品用柱层析方法提纯,洗脱比例为:二氯甲烷∶甲醇=30∶1,收集洗脱液共820ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出。得到淡粉色固体2.31g,产率:58.89%,即得到化合物h中间体-1。(2)向250ml的三口瓶中加入40mln-甲基吡咯烷酮,搅拌下依次加入2.00g化合物h中间体-1,2.05g吡啶,氮气保护,降温至0~10℃,开始滴加3.04g氯甲酸苯酯,滴毕,继续搅拌20~30分钟;取样tlc检测(甲醇∶二氯甲烷=1∶10)原料消失。加水40ml,有大量固体析出,继续搅拌30分钟。过滤,抽干,60℃鼓风干燥30分钟,得到棕黄色粉末2.21g,收率:79.54%,直接用于下一步化合物h的合成。(3)向100ml的三口瓶中加入25mln-甲基吡咯烷酮,搅拌下加入2.21g上述中间体。氮气保护,降温至0~10℃,开始滴加0.65g环丙胺,滴毕,继续搅拌20~30分钟;取样tlc检测(甲醇∶二氯甲烷=1∶10)原料消失。加水50ml,有大量固体析出,继续搅拌30分钟。过滤,抽干,40℃鼓风干燥30分钟,得到粗品用柱层析方法提纯,洗脱比例为::甲醇∶二氯甲烷=1∶20,收集洗脱液共530ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出。得到淡粉色固体0.89g,即得到化合物h,产率:44.04%,hplc纯度:99.60%。杂质h的1hnmr和esi-hrms谱图分别见附图21-22。实施例12:杂质i的合成向250ml的三口瓶中加入3.00glvtn-2,60mln-甲基吡咯烷酮。控温0~10℃,加入1.47g甲胺甲醇溶液,继续搅拌30分钟,tlc监测(二氯甲烷∶甲醇=10∶1),反应结束。加入33%丙酮水90ml,有大量固体产生,搅拌30分钟后,过滤,60℃鼓风干燥,得到化合物i粗品。将该粗品进行柱层析纯化,洗脱比例为:甲醇∶二氯甲烷=1∶20,收集洗脱液共760ml,控温30~40℃,真空度:-0.08mpa,减压蒸馏,蒸除溶剂至无馏分蒸出,得淡粉色固体0.90g,收率:34.70%,,即得到化合物i,hplc纯度:98.77%。杂质i的1hnmr和esi-hrms谱图分别见附图23-24。实施例13:甲磺酸乐伐替尼或乐伐替尼有关物质的hplc分析利用杂质外标法(2010年版中国药典),分析甲磺酸乐伐替尼或乐伐替尼中有关物质,并用外标法对各杂质进行定量。杂质的定量按照外标法进行计算,具体参见中国药典2010版二部附录vd。用于进行所述分析的方法为根据本发明的梯度hplc方法。使用的色谱条件如下:色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.08m醋酸铵溶液(醋酸调ph至3.5)(a),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:20℃流速:0.3ml/min梯度设计如下所述:样品配制:制备杂质定位溶液(1)化合物a~i、lvtn-1标准定位溶液:分别取化合物a~i及lvtn-1各约5mg,精密称定,分别置100ml容量瓶中,加乙腈或dmso超声使溶解,并用乙腈稀释至刻度,摇匀,制成浓度约为50μg/ml的溶液,作为化合物a~i及lvtn-1的贮备溶液。(2)加标混合溶液:取上述制备的甲磺酸乐伐替尼原料药适量(约含乐伐替尼25mg),精密称定,置50ml量瓶中,再精密加入化合物a~i、lvtn-1贮备溶液各0.5ml,加稀释剂适量,超声使溶解,再用稀释剂定容,摇匀,制成每1ml含乐伐替尼约0.5mg、含化合物a~i、lvtn-1分别约0.5μg的混合溶液,作为加标混合溶液。(3)制备化合物a~i、lvtn-1参比标准溶液或对照品溶液:精密量取化合物a~i贮备溶液0.5ml,置50ml容量瓶中,用稀释剂定容,摇匀,制成每1ml约含化合物a~i分别为0.5μg的溶液,作为化合物a~i的参比标准溶液或对照品溶液。(4)制备乐伐替尼供试品溶液:取实施例3制备的甲磺酸乐伐替尼原料药适量,约含乐伐替尼5mg,精密称定,置10ml容量瓶中,加稀释剂适量,超声使溶解,并用稀释剂定容,摇匀,既得。乐伐替尼原料药供试品溶液的浓度约为0.5mg/ml。(5)按照本发明专利实施例3制备的甲磺酸乐伐替尼和原研的甲磺酸乐伐替尼胶囊供试品溶液的制备:取甲磺酸乐伐替尼胶囊(lenvima购自eisaiinc.,10mg,批号:l15044)内容物研细粉末适量(约含乐伐替尼5mg),精密称定,置10ml容量瓶中,加稀释剂适量,超声使溶解,并用稀释剂定容,摇匀,用0.22μm有机针式过滤器过滤,弃去初滤液2ml,取续滤液,作为甲磺酸乐伐替尼胶囊供试品溶液。甲磺酸乐伐替尼胶囊供试品溶液的浓度约为0.5mg/ml。实施例14:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.02m醋酸铵溶液(a)(醋酸调ph至6.5),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:25℃流速:0.9ml/min实施例15:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.02m醋酸铵溶液(a)(醋酸调ph至5.0),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:35℃流速:0.5ml/min实施例16:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.03m醋酸铵溶液(a)(醋酸调ph至5.0),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:25℃流速:0.4ml/min实施例17:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.08m醋酸铵溶液(a)(醋酸调ph至5.0),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:35℃流速:0.3ml/min实施例18:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.03m醋酸铵溶液(a)(醋酸调ph至5.5),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:30℃流速:0.4ml/min实施例19:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.03m醋酸铵溶液(a)(醋酸调ph至5.0),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:35℃流速:0.3ml/min实施例20:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.03m醋酸铵溶液-(a)(醋酸调ph至4.5),乙腈(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:30℃流速:0.5ml/min实施例21:甲磺酸乐伐替尼的制备同实施例3,样品配制同实施例13,梯度设计同实施例13。色谱条件:色谱柱:watersxbridgebehshieldrp184.6×150mm,2.5μm或agilentporoshellhph-c184.6×150mm,2.7μm供试品溶液的浓度:0.5mg/ml参比标准溶液各杂质的浓度:1000ppm流动相:0.02m醋酸铵溶液(a)(醋酸调ph至3.5),乙腈-甲醇(b)检测波长:252nm稀释剂:二甲基亚砜∶乙腈∶a(2∶4∶4)柱温:25℃流速:1.0ml/min试验步骤:(1)在本发明梯度hplc方法下,在agilent1260hplc仪器(美国agilent公司)上,按照实施例13的色谱条件,取化合物a~i、lvtn-1定位溶液、加标混合溶液、化合物a~i、lvtn-1参比溶液、甲磺酸乐伐替尼供试品溶液、甲磺酸乐伐替尼胶囊供试品溶液各5μl,注入液相色谱仪,记录色谱图。(2)在本发明梯度hplc方法下,按照实施例13~21的色谱条件,取加标混合溶液5μl,注入液相色谱仪,记录色谱图。试验结果:化合物a~i、lvtn-1主成分甲磺酸乐伐替尼在本实施例13中梯度hplc方法下的定位结果见表3;在本发明实施例13~21的色谱条件下,加标混合溶液中各杂质之间的分离度、各杂质与甲磺酸乐伐替尼峰的相对保留时间见表4。利用实施例16的色谱条件,按照外标法计算供试品溶液中含有的化合物a~i、lvtn-1的含量及总杂质量,结果见表5,加标混合溶液的hplc谱图、甲磺酸乐伐替尼供试品溶液的hplc谱图、甲磺酸乐伐替尼胶囊、原研对照制剂lenvima-10mg规格的供试品溶液的hplc谱图分别见图25~图28,图25-28中的最高峰为乐伐替尼的峰,由lvtn表示。根据化合物a~i、lvtn-1及主成分定位结果及甲磺酸乐伐替尼和甲磺酸乐伐替尼胶囊有关物质的分析结果,甲磺酸乐伐替尼供试品溶液中检出了化合物c(相对保留时间约为0.47)、g(相对保留时间约为0.61)、h(相对保留时间约为0.78)、i(相对保留时间约为0.82)、b(相对保留时间约为0.96)和杂质a(相对保留时间约为1.12);甲磺酸乐伐替尼胶囊供试品溶液中检出了化合物c、g、h、i、b和4个未知杂质(归一化含量在0.01%以下的杂质忽略不计)。原研制剂lenvima胶囊中检出了杂质c、g、i、lvtn-1(相对保留时间约为0.94)、b、a和7个未知杂质(归一化含量在0.01%以下的杂质忽略不计)。表3化合物a~i、lvtn-1及甲磺酸乐伐替尼在实施例13方法中的定位及各峰之间的分离度表4实施例13~21条件下各杂质的相对保留时间注:色谱条件发生变化时,各杂质峰的保留时间会发生变化,但相对保留时间基本不变。杂质c和杂质e的分离度在实施例18的色谱条件下分离度最差为1.21,仍然大于难分离杂质要求的分离度不低于1.2。表5实施例3制备的甲磺酸乐伐替尼原料药和商购的lenvima胶囊中有关物质按照实施例16的色谱条件的检测结果n.d.代表“未检出”。需要说明的是在本发明中提及的所有文献在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,以上所述的是本发明的具体实施列及所运用的技术原理,在阅读了本发明的内容后,本领域技术人员可以对本发明做各种改动或修改而不背离本发明的精神与范围,这些等价形式同样落在本发明的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1