一种阿苯达唑及其代谢物的检测方法及应用与流程
本发明属于药物检测
技术领域:
,具体涉及一种阿苯达唑及其代谢物的检测方法及应用。
背景技术:
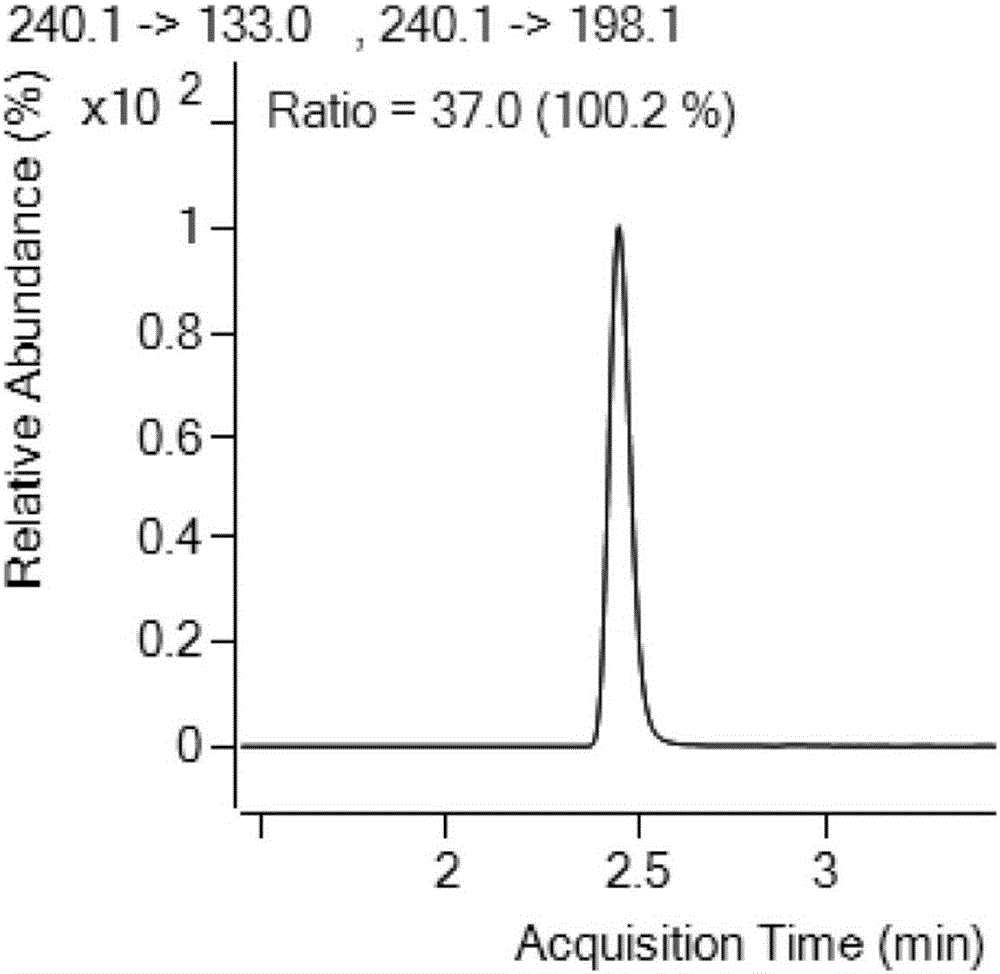
:目前,检测阿苯达唑的方法中使用乙醚、乙腈等毒性大的溶剂,或者直接使用乙腈提取,这些方法不仅使样品提取不充分,而且目标化合物在提取过程中极易遭到破坏,从而影响定量结果。技术实现要素:为了解决现有技术中的问题,本发明采取的技术方案是:一种阿苯达唑及其代谢物的检测方法,利用2,6-二叔丁基对甲酚和NaOH提取样品中的阿苯达唑及其代谢物,之后使用高效液相色谱质谱联用仪检测;所述2,6-二叔丁基对甲酚的浓度为1%;所述NaOH溶液的浓度为50%。在上述方案的基础上,所述阿苯达唑及其代谢物的检测方法步骤如下:1)提取纯化称取样品5.00g,加入5mL水、150μL50%NaOH溶液和1mL1%2,6-二叔丁基对甲酚,涡旋混合,再加入20mL乙酸乙酯,振荡1min超声提取20min,4000r/min离心5min,取上清液,向残渣中加入10mL乙酸乙酯重复提取一次,合并两次上清液于梨形瓶中,40℃旋蒸至干;向旋干后的梨形瓶中加入6mL乙腈饱和正己烷,振荡混匀,倒入离心管中,再向梨形瓶中加入2mL乙腈,涡旋超声1min后一并倒入离心管中,向离心管中加入6mL0.1mol/L的盐酸溶液,涡旋2min,4000r/min离心5min,除去正己烷层,备用;若乳化,超声并用吸管搅拌后离心;2)净化柱净化用5mL甲醇和5mL水平衡活化PCX固相萃取柱后,取提取液过PCX净化柱,分别用3mL水和3mL甲醇淋洗,然后用15mL体积浓度为5%氨水甲醇溶液洗脱,接收的洗脱液于40℃旋蒸至干后;分别加入1mL甲醇和1mL水,涡旋,过0.22μm尼龙滤膜,待测;3)高效液相色谱质谱联用仪检测质谱参数:离子源AJSESI源;GasTemp:325℃;GasFlow:10L/min;SheathGasTemp350℃;SheathGasflow:11L/min;液相参数:A为0.1%甲酸水溶液,B为甲醇。在上述方案的基础上,所述阿苯达唑及其代谢物的检测方法应用于检测动物源性食品中的阿苯达唑及其代谢物。在上述方案的基础上,所述动物源性食品为肌肉、蛋类、乳类、肝脏和肾脏。本发明的有益效果:本发明方法中用2,6-二叔丁基对甲酚和NaOH提取阿苯达唑及其代谢物,毒性较小;此外,阿苯达唑及其代谢物在水溶液中呈碱性,加入质量浓度为50%NaOH水溶液可使阿苯达唑及其代谢物成分子状态,利于有机试剂提取;加入质量浓度为1%2,6-二叔丁基对甲酚可以保护阿苯达唑不被氧化;乙酸乙酯为弱极性的有机试剂,其加入有利于阿苯达唑的提取且杂质提取的少;乙腈饱和正己烷用来去除样品中的油脂类杂质,加入盐酸可使阿苯达唑成离子状态有利于PCX净化柱净化;5%氨水甲醇呈强碱性,洗脱时可将PCX净化柱吸附的阿苯达唑置换下来。本发明的方法检测范围较广,相比于其他方法的单一基质,本方法不仅仅适用于动物肌肉而且也适用于肝脏等复杂基质的检测,众所周知,动物肝脏含有很多蛋白、脂肪、糖类、维生素和矿物质,净化不干净对目标化合物回收影响很大;本发明的方法使用2,6-二叔丁基对甲酚、NaOH和乙酸乙酯作为提取液,毒性小,成本低,节约时间,又能有效地减少对杂质的提取,简化净化步骤;本方法采用安捷伦PCX固相萃取小柱,也可用国产PCX小柱代替,有效降低检测成本;采用高效液相色谱-串联质谱法进行检测,相比于单一的高效液相色谱,具有灵敏度高,重复性好,检测限低的优势。附图说明图1阿苯达唑-2-氨基砜保留时间的测定,其中纵坐标为响应,横坐标为保留时间。图2阿苯达唑-2-氨基砜定量定性离子明细,其中纵坐标为响应,横坐标为保留时间。图3阿苯达唑-2-氨基砜质谱图,其中纵坐标为响应,横坐标为m/z。图4阿苯达唑亚砜保留时间的测定,其中纵坐标为响应,横坐标为保留时间。图5阿苯达唑亚砜定量定性离子明细,其中纵坐标为响应,横坐标为保留时间。图6阿苯达唑亚砜质谱图,其中纵坐标为响应,横坐标为m/z。图7阿苯达唑砜保留时间的测定,其中纵坐标为响应,横坐标为保留时间。图8阿苯达唑砜定量定性离子明细,其中纵坐标为响应,横坐标为保留时间。图9阿苯达唑砜质谱图,其中纵坐标为响应,横坐标为m/z。图10阿苯达唑保留时间的测定,其中纵坐标为响应,横坐标为保留时间。图11阿苯达唑定量定性离子明细,其中纵坐标为响应,横坐标为保留时间。图12阿苯达唑质谱图,其中纵坐标为响应,横坐标为m/z。图13阿苯达唑-2-氨基砜的标准曲线,其中纵坐标为峰面积响应,横坐标为阿苯达唑-2-氨基砜浓度。图14阿苯达唑亚砜的标准曲线,其中纵坐标为峰面积响应,横坐标为阿苯达唑亚砜浓度。图15阿苯达唑砜的标准曲线,其中纵坐标为峰面积响应,横坐标为阿苯达唑砜浓度。图16阿苯达唑的标准曲线,其中纵坐标为峰面积响应,横坐标为阿苯达唑浓度。图17阿苯达唑及其代谢物定量离子S/N图,其中纵坐标为响应,横坐标为保留时间,从上到下依次为阿苯达唑-2-氨基砜、阿苯达唑亚砜、阿苯达唑砜、阿苯达唑。具体实施方式在本发明中所使用的术语,除非有另外说明,一般具有本领域普通技术人员通常理解的含义。下面结合具体实施例,并参照数据进一步详细的描述本发明。以下实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。实施例1.标准溶液的配制1.1储备液的配制分别称量10.00mg阿苯达唑、阿苯达唑-2-氨基砜、阿苯达唑亚砜和阿苯达唑砜的标准品,溶解到甲醇中,定容到10mL,作为1000mg/L的储备液(置于-18℃冷藏保存)。1.2标准中间液的配制分别准确量取1mL阿苯达唑、阿苯达唑-2-氨基砜、阿苯达唑亚砜和阿苯达唑砜标准品的储备液,溶解到甲醇中,定容到10mL,作为100mg/L的标准中间液(置于-18℃冷藏保存)。1.3标准工作曲线的配制用标准中间液配制成1、5、10、20、50、100μg/L的标液(用甲醇+水(v/v=1:1)定容),作为工作曲线用的标准溶液。2.样品处理2.1提取纯化称取样品5.00g,加入5mL水、150μL50%NaOH溶液和1mL1%2,6-二叔丁基对甲酚,涡旋混合,再加入20mL乙酸乙酯,振荡1min超声提取20min,4000r/min离心5min,取上清液,向残渣中加入10mL乙酸乙酯重复提取一次,合并两次上清液于梨形瓶中,40℃旋蒸至干;向旋干后的梨形瓶中加入6mL乙腈饱和正己烷,振荡混匀,倒入离心管中,再向梨形瓶中加入2mL乙腈,涡旋超声1min后一并倒入离心管中,向离心管中加入6mL0.1mol/L的盐酸溶液,涡旋2min,4000r/min离心5min,除去正己烷层,备用;若乳化,超声并用吸管搅拌后离心;2.2净化柱净化用5mL甲醇和5mL水平衡活化PCX固相萃取柱后,取提取液过PCX净化柱,分别用3mL水和3mL甲醇淋洗,然后用15mL体积浓度为5%氨水甲醇溶液洗脱,接收的洗脱液于40℃旋蒸至干后;分别加入1mL甲醇和1mL水,涡旋,过0.22μm尼龙滤膜,待测;3.高效液相色谱质谱联用仪检测使用的高效液相色谱质谱联用仪为AgilentLC-MSMS1290/6460;质谱参数:离子源AJSESI源;GasTemp:325℃;GasFlow:10L/min;SheathGasTemp350℃;SheathGasflow:11L/min。液相参数:A为0.1%甲酸水溶液,B为甲醇。流动相梯度如表1所示:表1流动相梯度时间甲醇浓度1.00202.001004.001004.01205.5204.结果及分析4.1质谱结果阿苯达唑及其代谢物的质谱结果如表2所示:表2质谱结果4.2专属性4.2.1阿苯达唑-2-氨基砜的专属性50ppb阿苯达唑-2-氨基砜通过C18色谱柱分离,确定保留时间。如图1所示,保留时间约为2.4min,且色谱的峰形较佳,无干扰峰出现,其中横坐标为保留时间,纵坐标为响应。阿苯达唑-2-氨基砜定量定性离子明细如图2所示,其中纵坐标为响应,横坐标为保留时间。如图3所示,1ppm阿苯达唑-2-氨基砜上机,通过质谱优化得到3个离子对,纵坐标为响应,横坐标为m/z。4.2.2阿苯达唑亚砜的专属性50ppb阿苯达唑亚砜通过C18色谱柱分离,确定保留时间。如图4所示,保留时间约为2.9min,且色谱的峰形较佳,无干扰峰出现,其中横坐标为保留时间,纵坐标为响应。阿苯达唑亚砜定量定性离子明细如图5所示,其中纵坐标为响应,横坐标为保留时间。如图6所示,1ppm阿苯达唑亚砜上机,通过质谱优化得到3个离子对,纵坐标为响应,横坐标为m/z。4.2.3阿苯达唑砜的专属性50ppb阿苯达唑砜通过C18色谱柱分离,确定保留时间。如图7所示,保留时间约为2.9min,且色谱的峰形较佳,无干扰峰出现。其中横坐标为保留时间,纵坐标为响应。阿苯达唑砜定量定性离子明细如图8所示,其中纵坐标为响应,横坐标为保留时间。如图9所示,1ppm阿苯达唑砜上机,通过质谱优化得到3个离子对,纵坐标为响应,横坐标为m/z。4.2.4阿苯达唑的专属性50ppb阿苯达唑砜通过C18色谱柱分离,确定保留时间。如图10所示,保留时间约为3.159min,且色谱的峰形较佳,无干扰峰出现,横坐标为保留时间,纵坐标为响应。阿苯达唑定量定性离子明细如图11所示,其中纵坐标为响应,横坐标为保留时间。如图12所示,1ppm阿苯达唑上机,通过质谱优化各得到3个离子对,纵坐标为响应,横坐标为m/z。4.3标准曲线4.3.1阿苯达唑-2-氨基砜的标准曲线精密吸取阿苯达唑-2-氨基砜标准品,用50%甲醇水溶液稀释成系列浓度为1,5,10,20,50,100ng/mL标准溶液,分别进样测定,以峰面积响应为纵坐标,阿苯达唑-2-氨基砜浓度为横坐标。如图13所示,得到阿苯达唑-2-氨基砜的回归方程为Y=1378.144404*X-594.187990,R2=0.9999,由相关系数可见,在1ng/mL~100ng/mL标准曲线线性范围内,UPLC-MSMS法测定线性关系良好,此标准曲线可以用于准确定量。4.3.2阿苯达唑亚砜的标准曲线精密吸取阿苯达唑亚砜标准品,用50%甲醇水溶液稀释成系列浓度为1,5,10,20,50,100ng/mL标准溶液,分别进样测定,以峰面积响应为纵坐标,阿苯达唑亚砜浓度为横坐标。如图14所示,得到阿苯达唑亚砜的回归方程为Y=519.827117*X-28.361658,R2=0.9998,由相关系数可见,在1ng/mL~100ng/mL标准曲线线性范围内,UPLC-MSMS法测定线性关系良好,此标准曲线可以用于准确定量。4.3.3阿苯达唑砜的标准曲线精密吸取阿苯达唑砜标准品,用50%甲醇水溶液稀释成系列浓度为1,5,10,20,50,100ng/mL标准溶液,分别进样测定,以峰面积响应为纵坐标,阿苯达唑砜浓度为横坐标。如图15所示,得到阿苯达唑砜的回归方程为Y=1384.923769*X-533.206880,R2=0.9998,由相关系数可见,在1ng/mL~100ng/mL标准曲线线性范围内,UPLC-MSMS法测定线性关系良好,此标准曲线可以用于准确定量。4.3.4阿苯达唑精密吸取阿苯达唑标准品,用50%甲醇水溶液稀释成系列浓度为1,5,10,20,50,100ng/mL标准溶液,分别进样测定,以峰面积响应为纵坐标,阿苯达唑浓度为横坐标。如图16所示,得到阿苯达唑的回归方程为Y=8382.358953*X-2437.446609,R2=0.9999,由相关系数可见,在1ng/mL~100ng/mL标准曲线线性范围内,UPLC-MSMS法测定线性关系良好,此标准曲线可以用于准确定量。4.4检出限在基质中添加相当于样品含有1.0μg/kg的阿苯达唑及其代谢物的标准品,经前处理上机检测,计算其信噪比(S/N)>10,如图17所示。测得本发明方法对阿苯达唑及其代谢物的检出限为1.0μg/kg。4.5验证基体及回收率基体及回收率的验证如表3所示:表3基体及回收率的验证从市场上购买牛肉、猪肝、猪肾、从超市购买鸡蛋、原料乳、鲫鱼基质做加标实验回收率70%-90%之间,说明本方法稳定性良好。5.结论通过运用UPLC-LCMSMS法对动物源基体中的阿苯达唑及其代谢物进行测定,确定使用2,6-二叔丁基对甲酚、NaOH和乙酸乙酯进行提取,正己烷纯化,PCX小柱进行净化,使阿苯达唑及其代谢物色谱峰达到基线分离,峰形良好,检测限低,灵敏度高,方法回收稳定,本研究为动物源基质中的阿苯达唑及其代谢物残留量的测定建立了一种可靠地分析方法。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1