一种9‑去甲基‑α‑二氢丁苯那嗪及其杂质的分离测定方法与流程
本发明属于药物分析领域,具体涉及一种9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法。
背景技术:
Ⅱ型囊泡单胺转运体(VMAT2)是一种位于神经元胞质内囊泡膜上的膜蛋白,在单胺类递质的重摄取过程中起着关键作用。分子克隆技术鉴定表明,VMAT2主要分布于人类及哺乳动物的中枢神经系统与胰脏组织内。VMAT2的变化与多种疾病有关,如帕金森病(PD)、阿尔茨海默病(AD)、亨廷顿病(HD)、糖尿病等。近年来,以VMAT2为靶标进行放射性示踪显像,对相关疾病进行早期诊断、分级、鉴别诊断、疗效监测研究,成为核医学研究的热点之一。到目前为止,国内外已成功应用于临床的VMAT2显像剂,都是用于正电子发射计算机断层(PET)成像的正电子示踪药物,如:11C-α-DTBZ、18F-FP-α-DTBZ等。
9-去甲基-α-二氢丁苯那嗪(9-DM-α-DTBZ)是制备PET显像药物11C-α-DTBZ的标记前体化合物,标记过程如下所示:
由于11C半衰期仅为20min,临床上使用示踪药物11C-α-DTBZ需要通过标记前体化合物9-DM-α-DTBZ现场制备而得。标记前体化合物9-DM-α- DTBZ中杂质的含量影响到PET显像药物11C-α-DTBZ的纯度和标记率,进一步影响最终的显像效果。
目前,已报道的9-DM-α-DTBZ常用的制备方法为:α-二氢丁苯那嗪(α-DTBZ)在氢化钠、六甲基磷酰三胺及N-甲基苯胺的作用下脱除甲基生成9-DM-α-DTBZ(Chirality,1997,9:59-62)。实验表明,反应完成后,反应液中的溶剂和反应原料氢化钠、六甲基磷酰三胺在反应后处理的过程中即可除去,而反应原料α-DTBZ及N-甲基苯胺则较难去除干净。因此,在9-DM-α-DTBZ的制备过程中,可能混有的主要杂质为α-DTBZ及N-甲基苯胺。
到目前为止,还没有关于9-DM-α-DTBZ及其杂质α-DTBZ和N-甲基苯胺的分离测定方法的相关报道。尽管有文献报道:HPLC法测定丁苯那嗪原料药含量,然而,一方面由于9-DM-α-DTBZ原料药所含杂质(主要为α-DTBZ和N-甲基苯胺)和丁苯那嗪原料药所含杂质的差别,另一方面由于9-DM-α-DTBZ和丁苯那嗪化学结构的差别,申请人通过研究发现,上述HPLC法测定丁苯那嗪原料药含量并不能用于9-DM-α-DTBZ及其杂质的分离测定,采用此HPLC法时9-DM-α-DTBZ与杂质不能完全分离、分离度较差,进而导致9-DM-α-DTBZ及其杂质α-DTBZ和N-甲基苯胺的含量无法进行测定,也就无法对9-DM-α-DTBZ的纯度和含量进行质量控制,难以确保PET显像药物11C-α-DTBZ的纯度和标记率,从而不利于PET显像药物11C-α-DTBZ使用的有效性和安全性。
因此,建立一种9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法具有重要的意义。
技术实现要素:
本发明要解决的第一个技术问题是现有技术无法实现9-去甲基-α-二氢丁苯那嗪原料药中的9-DM-α-DTBZ及其主要杂质α-DTBZ和N-甲基苯胺完全分离的问题,本发明要解决的第二个技术问题是现有技术无法实现9-去甲基-α-二氢丁苯那嗪原料药中的9-DM-α-DTBZ及其主要杂质α-DTBZ和N-甲基苯胺的含量同时进行测定的问题。
为解决上述技术问题,本发明提出一种9-去甲基-α-二氢丁苯那嗪及其杂质α-DTBZ和N-甲基苯胺的分离测定方法。
为解决上述技术问题,本发明是通过以下技术方案来实现的:
本发明提供一种9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为40~90:10~60的醋酸铵缓冲液-甲醇进行溶解,制成浓度为0.5~1.5mg/mL的溶液,作为供试品溶液;
照高效液相色谱法,以反相色谱柱为色谱柱,以体积比为40~90:10~60的醋酸铵缓冲液-甲醇为流动相,流速为0.5~1.5mL/min,检测波长为270~290nm,柱温为20~35℃;
取所述供试品溶液5~15μL,注入高效液相色谱仪,测定。
优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,以体积比为50~75:25~50的醋酸铵缓冲液-甲醇为流动相。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,
以体积比为70:30的醋酸铵缓冲液-甲醇为流动相;或者
以体积比为50:50的醋酸铵缓冲液-甲醇为流动相;或者
以体积比为75:25的醋酸铵缓冲液-甲醇为流动相。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,所述醋酸铵缓冲液的浓度为40~60mM、pH值为4~5。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,所述醋酸铵缓冲液的浓度为50mM、pH值为4.5。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,所述反相色谱柱的填充剂为十八烷基硅烷键合硅胶。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,所述反相色谱柱的填充剂为粒径5μm的十八烷基硅烷键合硅胶。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,所述反相色谱柱为5μm、4.6×150mm的Sunfire C18反相色谱柱。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为70:30的醋酸铵缓冲液-甲醇进行溶解,制成浓度为1mg/mL的溶液,作为供试品溶液;
照高效液相色谱法,以5μm、4.6×150mm的Sunfire C18反相色谱柱为色谱柱,以体积比为70:30的醋酸铵缓冲液-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;所述醋酸铵缓冲液的浓度为50mM、pH值为4.5;
取所述供试品溶液10μL,注入高效液相色谱仪,测定;或者
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为50:50的醋酸铵缓冲液-甲醇进行溶解,制成浓度为1.0mg/mL的溶液,作为供试品溶液;
照高效液相色谱法,以5μm、4.6×150mm的Sunfire C18反相色谱柱为色谱柱,以体积比为50:50的醋酸铵缓冲液-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;所述醋酸铵缓冲液的浓度为50mM、pH值为4.5;
取所述供试品溶液10μL,注入高效液相色谱仪,测定;或者
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为75:25的醋酸铵缓冲液-甲醇进行溶解,制成浓度为1mg/mL的溶液,作为供试品溶液;
照高效液相色谱法,以5μm、4.6×150mm的Sunfire C18反相色谱柱为色谱柱,以体积比为75:25的醋酸铵缓冲液-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;所述醋酸铵缓冲液的浓度为50mM、pH值为4.5;
取所述供试品溶液10μL,注入高效液相色谱仪,测定。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,还包括如下9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液、N-甲基苯胺对照品溶液的制备和测定的步骤:
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为0.5~1.5mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为0.5~1.5mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为0.5~1.5mg/mL的溶液,作为N-甲基苯胺对照品溶液;
分别取所述9-去甲基-α-二氢丁苯那嗪对照品溶液、所述α-二氢丁苯那嗪对照品溶液和所述N-甲基苯胺对照品溶液各5~15μL,注入高效液相色谱仪,测定。
进一步优选地,本发明上述9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,还包括如下9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液、N-甲基苯胺对照品溶液的制备和测定的步骤:
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
分别取所述9-去甲基-α-二氢丁苯那嗪对照品溶液、所述α-二氢丁苯那嗪对照品溶液和所述N-甲基苯胺对照品溶液各10μL,注入高效液相色谱仪,测定。
与现有技术相比,本发明的上述技术方案具有以下优点:
本发明9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,以5μm、4.6×150mm的Sunfire C18反相色谱柱为色谱柱,以体积比为50~75:25~50的醋酸铵缓冲液-甲醇为流动相,不仅可以使9-去甲基-α-二氢丁苯那嗪原料药中9-DM-α-DTBZ及其杂质α-DTBZ和N-甲基苯胺实现完全分离,而且可以对9-DM-α-DTBZ及其杂质α-DTBZ和N-甲基苯胺的含量同时进行测定,为9-去甲基-α-二氢丁苯那嗪原料药中9-去甲基-α-二氢丁苯那嗪的含量测定和有关物质的含量测定提供了一种简便、快速、灵敏的检测方法,从而能够实现对9-DM-α-DTBZ原料药进行质量控制,进而确保PET显像药物11C-α-DTBZ的纯度和标记率,有利于PET显像药物11C-α-DTBZ使用的有效性和安全性。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
图1为实施例1中供试品溶液的HPLC色谱图;
图2为实施例1中9-去甲基-α-二氢丁苯那嗪对照品的HPLC色谱图;
图3为实施例1中α-二氢丁苯那嗪对照品的HPLC色谱图;
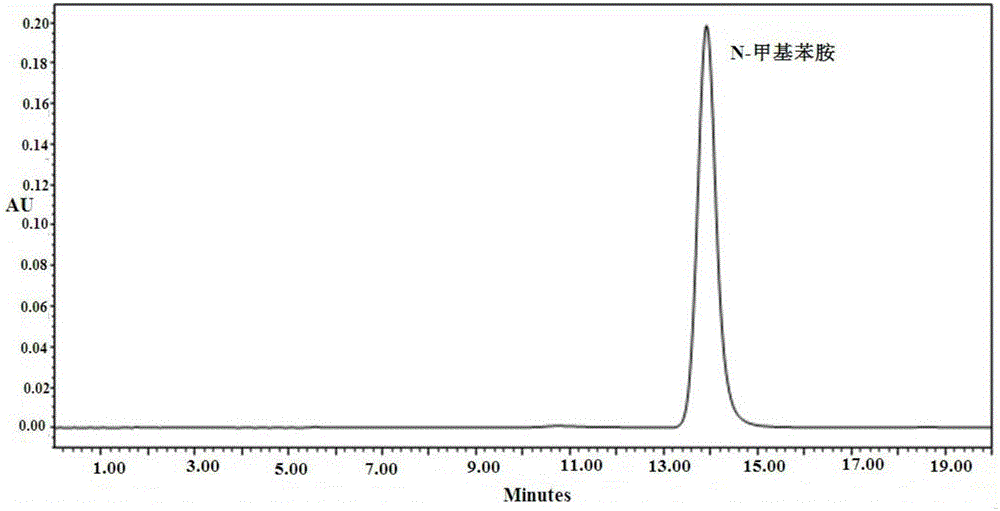
图4为实施例1中N-甲基苯胺对照品的HPLC色谱图;
图5为实施例2中供试品溶液的HPLC色谱图;
图6为实施例3中供试品溶液的HPLC色谱图;
图7为对比例1中供试品溶液的HPLC色谱图;
图8为对比例2中供试品溶液的HPLC色谱图;
图9为对比例3中供试品溶液的HPLC色谱图;
图1-9中,9-DM-α-DTBZ表示的为9-去甲基-α-二氢丁苯那嗪,α-DTBZ表示的为α-二氢丁苯那嗪。
具体实施方式
本发明以下实施例和实验例中,9-DM-α-DTBZ表示的为9-去甲基-α-二氢丁苯那嗪,α-DTBZ表示的为α-二氢丁苯那嗪。
实施例1
本实施例9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为70:30的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇进行溶解,制成浓度为1mg/mL的溶液,作为供试品溶液;
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
照高效液相色谱法,以Sunfire C18反相色谱柱(5μm、4.6×150mm)为色谱柱,以体积比为70:30的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;
分别取供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液各10μL,注入高效液相色谱仪,测定。
本实施例中供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液的HPLC色谱图分别如图1-4所示。
由图1-4可知,实施例1中,9-DM-α-DTBZ与反应原料α-DTBZ、N-甲基苯胺的保留时间差异大,三者完全分离,分离效果良好;9-DM-α-DTBZ的保留时间为6.22min,含量为65.1%;α-DTBZ的保留时间为10.60min,含量为16.6%;N-甲基苯胺的保留时间为13.88min,含量为18.3%。
实施例2
本实施例9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为50:50的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇进行溶解,制成浓度为1.0mg/mL的溶液,作为供试品溶液;
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
照高效液相色谱法,以Sunfire C18反相色谱柱(5μm、4.6×150mm)为色谱柱,以体积比为50:50的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;
分别取供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液各10μL,注入高效液相色谱仪,测定。
本实施例中供试品溶液的HPLC色谱图分别如图5所示。
由图5可知,实施例2中,9-DM-α-DTBZ与反应原料α-DTBZ分离效果稍差;9-DM-α-DTBZ的保留时间为2.45min,含量为65.4%;α-DTBZ的保留时间为3.02min,含量为15.4%;N-甲基苯胺的保留时间为6.68min,含量为19.2%。
实施例3
本实施例9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为75:25的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇进行溶解,制成浓度为1mg/mL的溶液,作为供试品溶液;
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
照高效液相色谱法,以Sunfire C18反相色谱柱(5μm、4.6×150mm)为色谱柱,以体积比为75:25的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-甲醇为流动相,流速为1mL/min,检测波长为280nm,柱温为25℃;
分别取供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液各10μL,注入高效液相色谱仪,测定。
本实施例中供试品溶液的HPLC色谱图分别如图6所示。
由图6可知,实施例3中,9-DM-α-DTBZ与反应原料α-DTBZ、N-甲基苯胺的保留时间差异较大,三者完全分离,分离效果较好;9-DM-α-DTBZ的保留时间为9.65min,含量为66.2%;α-DTBZ的保留时间为16.28min,含量为16.7%;N-甲基苯胺的保留时间为18.79min,含量为17.1%。
实施例4
本实施例9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为90:10的醋酸铵缓冲液(60mM、pH值为4)-甲醇进行溶解,制成浓度为1.5mg/mL的溶液,作为供试品溶液;
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
照高效液相色谱法,以Sunfire C18反相色谱柱(5μm、4.6×150mm)为色谱柱,以体积比为90:10的醋酸铵缓冲液(60mM、pH值为4)-甲醇为流动相,流速为1.5mL/min,检测波长为270nm,柱温为35℃;
分别取供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液各5μL,注入高效液相色谱仪,测定。
实施例5
本实施例9-去甲基-α-二氢丁苯那嗪及其杂质的分离测定方法,包括以下步骤:
称取待测9-去甲基-α-二氢丁苯那嗪原料药适量,加入体积比为40:60的醋酸铵缓冲液(40mM、pH值为5)-甲醇进行溶解,制成浓度为0.5mg/mL的溶液,作为供试品溶液;
取9-去甲基-α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为9-去甲基-α-二氢丁苯那嗪对照品溶液;
取α-二氢丁苯那嗪的对照品适量,制成浓度为1mg/mL的溶液,作为α-二氢丁苯那嗪对照品溶液;
取N-甲基苯胺的对照品适量,制成浓度为1mg/mL的溶液,作为N-甲基苯胺对照品溶液;
照高效液相色谱法,以Sunfire C18反相色谱柱(5μm、4.6×150mm)为色谱柱,以体积比为40:60的醋酸铵缓冲液(40mM、pH值为5)-甲醇为流动相,流速为0.5mL/min,检测波长为290nm,柱温为20℃;
分别取供试品溶液、9-去甲基-α-二氢丁苯那嗪对照品溶液、α-二氢丁苯那嗪对照品溶液和N-甲基苯胺对照品溶液各15μL,注入高效液相色谱仪,测定。
实施例6
本实施例与实施例1的区别仅在于:流速为0.8mL/min;其余实验条件和实验操作步骤均与实施例1相同。
实施例7
本实施例与实施例1的区别仅在于:流速为1.2mL/min;其余实验条件和实验操作步骤均与实施例1相同。
对比例1
本对比例与实施例1的区别仅在于:制备供试品溶液的步骤中,加入体积比为20:80的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈进行溶解;作为供试品溶液以体积比为20:80的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈为流动相;以Lichrospher C18反相色谱柱(5μm、4.6×250mm)为色谱柱;流速为0.8mL/min;其余实验条件和实验操作步骤均与实施例1相同。
本对比例中供试品溶液的HPLC色谱图分别如图7所示。
由图7可知,对比例1中,9-DM-α-DTBZ与反应原料α-DTBZ的保留时间基本一致,峰形几乎重叠在一起,分离效果差;9-DM-α-DTBZ的保留时间为2.10min,α-DTBZ的保留时间为2.19min,N-甲基苯胺的保留时间为3.00min。
对比例2
本对比例与对比例1的区别仅在于:制备供试品溶液的步骤中,加入体积比为50:50的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈进行溶解;作为供试品溶液以体积比为50:50的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈为流动相;其余实验条件和实验操作步骤均与对比例1相同。
本对比例中供试品溶液的HPLC色谱图分别如图8所示。
由图8可知,对比例2中,9-DM-α-DTBZ与反应原料α-DTBZ的分离度为0.7,分离效果差;9-DM-α-DTBZ的保留时间为2.24min,α-DTBZ的保留时间为1.73min,N-甲基苯胺的保留时间为6.93min。
对比例3
本对比例与对比例1的区别仅在于:制备供试品溶液的步骤中,加入体积比为80:20的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈进行溶解;作为供试品溶液以体积比为80:20的醋酸铵缓冲液(浓度为50mM、pH值为4.5)-乙腈为流动相;其余实验条件和实验操作步骤均与对比例1相同。
本对比例中供试品溶液的HPLC色谱图分别如图9所示。
由图9可知,对比例3中,9-DM-α-DTBZ与反应原料α-DTBZ的峰形完全重叠在一起,分离效果差;9-DM-α-DTBZ的保留时间为6.60min,α-DTBZ的保留时间为6.50min,N-甲基苯胺的保留时间为9.96min。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!