生姜及其炮制品中姜辣素成分的检测方法与流程
本发明属于中药成分检测技术领域,涉及一种生姜及其炮制品中姜辣素成分的检测方法。
背景技术:
生姜为姜科植物姜zingiberofficinalerosc.的新鲜根茎,其传统炮制品有干姜、煨姜、炮姜、姜炭等,系分别采用干燥、煨制(包裹烫)、以沙(土)烫炒至一定程度、炒炭等所得。《日本药典》记载有采用蒸或煮制的方法炮制生姜,其称为亁姜。
生姜的化学成分包含多糖、生姜蛋白、黄酮类成分、姜辣素类成分、挥发油类成分。而姜辣素类成分(结构中包含3-甲氧基-4-羟基苯基官能团)作为生姜中公认的活性成分,其类型包括姜酚、姜烯酚和姜酮等。
常用的中药的定量评价方法有理化分析的方法如滴定法、重量法等;仪器分析的方法如紫外分光光度法、高效液相色谱法、气相色谱法等,而仪器分析的方法在实现准确定量时均需要使用标准物质(对照品),一些对照品由于自身化学性状的稳定性、含量等因素导致其难以获得、价格高,进行多成分的测定时其检测成本尤高。采用仪器分析的方法实现含量测定时,有两个关键因素,一是取得保留时间实现对物质的色谱峰进行定位,二是取得对应物质的响应值(仪器信号强度,如峰面积、峰高等,与物质实际浓度的关系)实现定量的目的。
目前色谱峰的定位方法有对照品直接定位法,该方法是目前定位最为准确的方法,但由于对照品价格昂贵,进行多成分测定时价格尤高。对照提取物法,但是对照提取物的制备本身不易,使用前还需要进行对照提取物中各成分的标定,虽然能够实现多成分的测定,但应用中成本亦较高。
一测多评法使用的相对保留时间法、线性回归色谱峰校正法、保留时间差法等间接定位的方法,通过计算进行色谱峰的定位,但是这些方法均存在误差,在应用中受到色谱柱、测定仪器、流动相等硬件的影响较大。同时,姜类产品色谱图中色谱峰较为丰富,通过计算得到的保留时间附近往往有多个色谱峰,该方法在实际应用中亦存在问题。
技术实现要素:
本发明的目的在于:针对现有技术存在的问题,提供一种生姜及其炮制品中姜辣素成分的检测方法,能以1-3种对照品实现多种姜辣素成分的色谱峰定位检测,且定位准确,成本低。
为了实现上述目的,本发明采用的技术方案为:
一种生姜及其炮制品中姜辣素成分的检测方法,包括以下步骤:
(1)取6-姜酚、8-姜酚和10-姜酚对照品中的一种或几种,置于容器中,然后加热;
(2)将步骤(1)加热后的容器取出,冷却后加入溶剂溶解,得到同时含有姜酮、姜酚及姜烯酚的定位用混合对照品溶液;
(3)将步骤(2)得到的混合对照品溶液注入色谱仪,在色谱图上出峰,利用该色谱图可实现生姜及其炮制品中姜辣素成分的色谱峰定位检测。
作为本发明的优选方案,所述步骤(1)中,加热前先往容器中加水并密闭,加入的水的质量与对照品的总质量之比为1:1-10。
作为本发明的优选方案,所述步骤(1)中,加热方式为烘箱烘制、红外加热或微波加热。
作为本发明的优选方案,所述步骤(1)中,加热温度为60-220℃,加热时间为0.5-4h。
作为本发明的优选方案,所述步骤(1)中,加热温度为120-140℃,加热时间为2-4h。
作为本发明的优选方案,所述步骤(2)中,加入的溶剂为乙腈。
本发明的有益效果在于:
本发明利用生姜中成分本身的化学性质,通过简单的工艺促进姜酚类成分转变为姜烯酚及姜酮,以1-3种对照品实现多种姜辣素成分的色谱峰定位检测,且相对于一测多评法定位更加准确,而相对于对照品直接定位法,定位同等准确,但减少了对照品的种类,成本更低。
因此,本发明准确、低成本的实现了生姜及其炮制品中姜辣素成分的色谱峰定位检测,能节约生姜及其炮制品质量控制的成本,方便快捷,环保经济,应用广泛。
附图说明
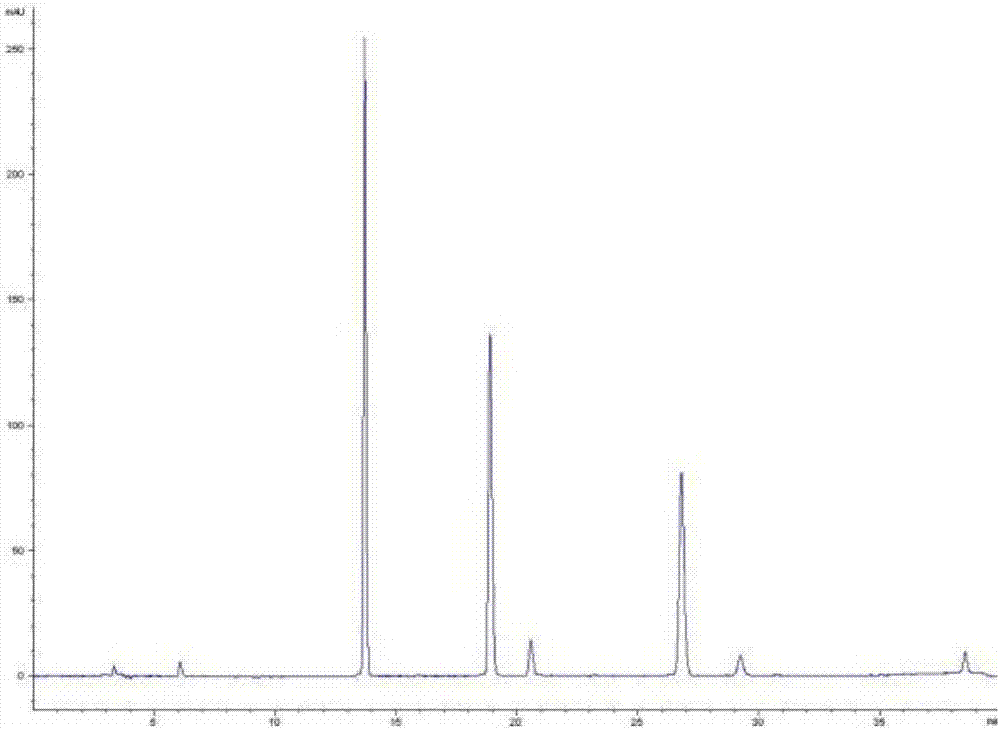
图1为仅将6-姜酚、8-姜酚和10-姜酚混合加乙腈得到混合对照品溶液,未经过实施例1处理而直接进样分析得到的色谱图;
图2为实施例1得到的色谱图;
图3为对比例1得到的色谱图。
具体实施方式
为了使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明的优选实施例进行详细的描述。
本发明中,姜辣素成分包括姜酮(英文名称zingerone)、6-姜酚(英文名称6-gingerol)、8-姜酚(英文名称8-gingerol)、10-姜酚(英文名称10-gingerol)、6-姜烯酚(英文名称6-shogaol)、8-姜烯酚(英文名称8-shogaol)和10-姜烯酚(英文名称10-shogaol)。
以下实施例与对比例的色谱条件为:
色谱柱:watersbridgetmc18(250×4.6nm,5μm);流动相为a相:0.2%甲酸-水,b相:0.2%甲酸-乙腈;采用二元梯度洗脱,洗脱程序为0-3min,b相60%~60%;3-8min,b相60%-50%;8-10min,b相50%~40%;10-20min,b相40%~32%;20-30min,b相32%~25%;30-35min,b相25%~5%;35-40min,b相5%~5%;流速:0.8ml/min;检测波长:280nm;柱温:30℃;进样量:10μl。
实施例1
(1)取6-姜酚、8-姜酚和10-姜酚对照品各0.1mg置于容器中,然后加0.1μl水密闭,置于烘箱中120℃烘制4h;
(2)将步骤(1)烘制后的容器取出,冷却后加入乙腈溶解,得到同时含有姜酮、6-姜酚、8-姜酚、10-姜酚、6-姜烯酚、8-姜烯酚和10-姜烯酚的定位用混合对照品溶液;
(3)将步骤(2)得到的混合对照品溶液在前述色谱条件下进样分析,得到如表4所示的各成分保留时间和如图2所示的色谱图。
对比例1对照品直接定位法
(1)精密称定姜酮、6-姜酚、8-姜酚、10-姜酚、6-姜烯酚、8-姜烯酚、10-姜烯酚适量,加乙腈分别制成每1ml各含0.1mg的溶液,混合后得到混合对照品溶液;
(2)将步骤(1)得到的混合对照品溶液在前述色谱条件下进样分析,得到如表4所示的各成分保留时间和如图3所示的色谱图。
对比例2相对保留时间法
(1)测定7种姜辣素成分彼此相对保留时间(相对保留时间=6品牌色谱柱的横坐标成分的平均保留时间/6品牌色谱柱的纵坐标成分的平均保留时间),如下表1所示:
(2)精密称定6-姜酚适量,加乙腈制成每1ml含0.1mg的溶液,得到6-姜酚对照品溶液;
(3)将步骤(2)得到的6-姜酚对照品溶液在前述色谱条件下进样分析,并用以下公式计算各成分保留时间:待测成分的保留时间tx=ft×t,其中tx为待测成分的保留时间,ft为待测成分/对照成分的相对保留时间,t为对照成分的保留时间;得到如表4所示的各成分保留时间。
对比例3保留时间差法
(1)测定7种姜辣素成分的保留时间差,如下表2和表3所示:
(2)精密称定6-姜酚适量,加乙腈制成每1ml含0.1mg的溶液,得到6-姜酚对照品溶液;
(3)将步骤(2)得到的6-姜酚对照品溶液在前述色谱条件下进样分析,并用以下公式计算各成分保留时间:待测成分的保留时间tx=t+δt,其中tx为待测成分的保留时间,t为对照成分的保留时间,δt为保留时间差;得到如表4所示的各成分保留时间。
图1为仅将6-姜酚、8-姜酚和10-姜酚混合加乙腈得到混合对照品溶液,未经过实施例1处理而直接进样分析得到的色谱图。
通过图1、图2和图3的对比可知,经过实施例1处理,能够把6-姜酚、8-姜酚、10-姜酚一次性转变为姜酮、6-姜烯酚、8-姜烯酚以及10-姜烯酚,同时保留原有的6-姜酚、8-姜酚、10-姜酚,因此在图1的3种成分色谱峰的基础上,增加了4种成分的色谱峰,7种成分在色谱图上出峰顺序依次为姜酮、6-姜酚、8-姜酚、6-姜烯酚、10-姜酚、8-姜烯酚、10-姜烯酚,各成分出峰顺序和位置与图3的对照品直接定位得到的色谱图完全一致,且不出现明显杂质,可见,实施例1能以3种对照品实现7种姜辣素成分的色谱峰准确定位检测。
表4的结果表明,实施例1的方法得到的7种姜辣素成分保留时间与对比例1的对照品直接定位法得到的7种姜辣素成分保留时间一致,而一测多评法(对比例2的相对保留时间法、对比例3的保留时间差法等间接定位的方法)的数据存在一定的偏差,一测多评法对姜酮、8-姜烯酚、10-姜烯酚等成分定位时无法准确分辨。
综上,实施例1以3种对照品实现7种姜辣素成分的色谱峰定位检测,且相对于一测多评法定位更加准确,而相对于对照品直接定位法,定位同等准确,但减少了对照品的种类,成本更低。
在实施例1的7种姜辣素成分色谱峰定位检测的基础上,结合常规的一测多评的定量方法,根据相对校正因子,通过计算,即可以实现7种姜辣素成分的定量测定。如表5和表6所示,与对照品直接测定结果相比,准确度较高。
本发明是以1-3种对照品实现多种姜辣素成分的色谱峰定位检测,不局限于实施例1的以3种对照品实现7种姜辣素成分的色谱峰定位检测,本发明也可以是1种或2种对照品实现多种姜辣素成分的色谱峰定位检测,如单独使用6-姜酚即可实现对6-姜酚、6-姜烯酚、姜酮的定位检测。另外,同时本发明亦不局限于在液相系统的应用,该方法原理在其他色谱系统中亦是可行的。
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管通过参照本发明的优选实施例已经对本发明进行了描述,但本领域的普通技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离所附权利要求书所限定的本发明的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!