一种清火片的质量控制方法与流程
本发明属于医药技术领域,具体涉及一种清火片,特别涉及一种清火片的质量控制方法。
背景技术:
清火片由大青叶,大黄,石膏,薄荷脑这4味药材组成,是中医用于治疗清热泻火,通便、咽喉肿痛,牙痛,头目眩晕,口鼻生疮,风火目赤,大便不通的要药。清火片属于中成药,是以中草药为原料经制剂加工制成各种不同剂型的中药制品,常规的质量控制方法为显微鉴别、薄层鉴别和单一指标成分类型的含量测定,无法准确而全面的控制产品质量,中药制剂的多种成分及各种赋形剂会对质量检测带来多方面的干扰,样品的前处理程序若不进行科学的验证很可能会造成不准确的试验结果。所以寻找灵敏、准确、快捷和全面控制质量的检测方法,成为中药质量控制的关键。现行的标准(国家食药监局标准ybz04782008),仅对方中大黄的大黄素与大黄酚两个成分进行含量测定,而明显忽略了芦荟大黄素、大黄酸、和大黄素甲醚的监控。
公开于该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域一般技术人员所公知的现有技术。
技术实现要素:
本发明的目的在于提供一种清火片的质量控制方法,来克服现有技术中存在的缺陷。
为实现上述目的,本发明提供了一种清火片的质量控制方法,采用高效液相法同时测定清火片中游离蒽醌与结合蒽醌的含量,包括以下步骤:
1)色谱条件:以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%-0.5%磷酸溶液为流动相b,梯度洗脱,0→15min,60%→70%a,40%→30%b;15→17min,60%→82%a,40%→18%b;17→30min,75%→82%a,25%→18%b,检测波长为254nm;
2)对照品的溶液的制备:精密称取大黄素对照品适量,加甲醇制成每1ml含大黄素10μg的溶液,即得;
3)游离蒽醌供试品溶液的制备:本品除去包衣研细,取约0.4g,称定重量后加入适量甲醇,称定总重量,加热回流30-90min,冷却后称定总重量,用甲醇补足减失的重量,摇匀,取续滤液即得,余下续滤液备用;
4)总蒽醌供试品溶液的制备:定量吸取步骤(3)最后备用的续滤液并挥去甲醇,加2-4倍体积的8%盐酸溶液混匀,加适量三氯甲烷,加热回流30-60min,分取三氯甲烷层,减压回收溶剂至干,残渣加甲醇适量使溶解并过滤,取续滤液即得;
5)游离蒽醌含量测定法:分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪测定,并按下式计算即得:游离蒽醌含量(mg/片)=(芦荟大黄素峰面积*0.716+大黄酸峰面积*0.874+大黄素峰面积*1.00+大黄酚峰面积*0.688+大黄素甲醚峰面积*1.007)*(大黄素对照品浓度/大黄素对照品峰面积)*稀释倍数*平均片重/称样量;
6)总蒽醌含量测定法:分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪测定,并按下式计算即得:总蒽醌含量(mg/片)=(芦荟大黄素峰面积*0.716+大黄酸峰面积*0.874+大黄素峰面积*1.00+大黄酚峰面积*0.688+大黄素甲醚峰面积*1.007)*(大黄素对照品浓度/大黄素对照品峰面积)*稀释倍数*平均片重/称样量。
优选地,上述技术方案中,步骤1具体为:色谱条件:以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%-0.3%磷酸溶液为流动相b,梯度洗脱,0→15min,70%a,30%b;15→17min,70%→82%a,30%→18%b;17→30min,82%a,18%b,检测波长为254nm。
优选地,上述技术方案中,步骤1具体为:色谱条件:以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.3%-0.5%磷酸溶液为流动相b,梯度洗脱,0→15min,65%a,35%b;15→17min,65%→80%a,35%→20%b;17→30min,80%a,20%b,检测波长为254nm。
优选地,上述技术方案中,步骤1具体为:色谱条件:以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.3%-0.5%磷酸溶液为流动相b,梯度洗脱,0→15min,60%a,40%b;15→17min,60%→75%a,40%→25%b;17→30min,75%a,25%b,检测波长为254nm。
优选地,上述技术方案中,步骤3具体为:游离蒽醌供试品溶液的制备:取本品20片,除去包衣,精密称定,研细,取约0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,加热回流30或60或90min,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,取续滤液即得,余下续滤液备用。
优选地,上述技术方案中,步骤4具体为:总蒽醌供试品溶液的制备:取步骤(3)最后备用的续滤液5ml,置烧瓶中,挥去甲醇,加8%盐酸溶液10ml或者20ml,超声处理5分钟,再加三氯甲烷10ml,加热回流1小时,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10ml,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇适量使溶解,转移至10ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
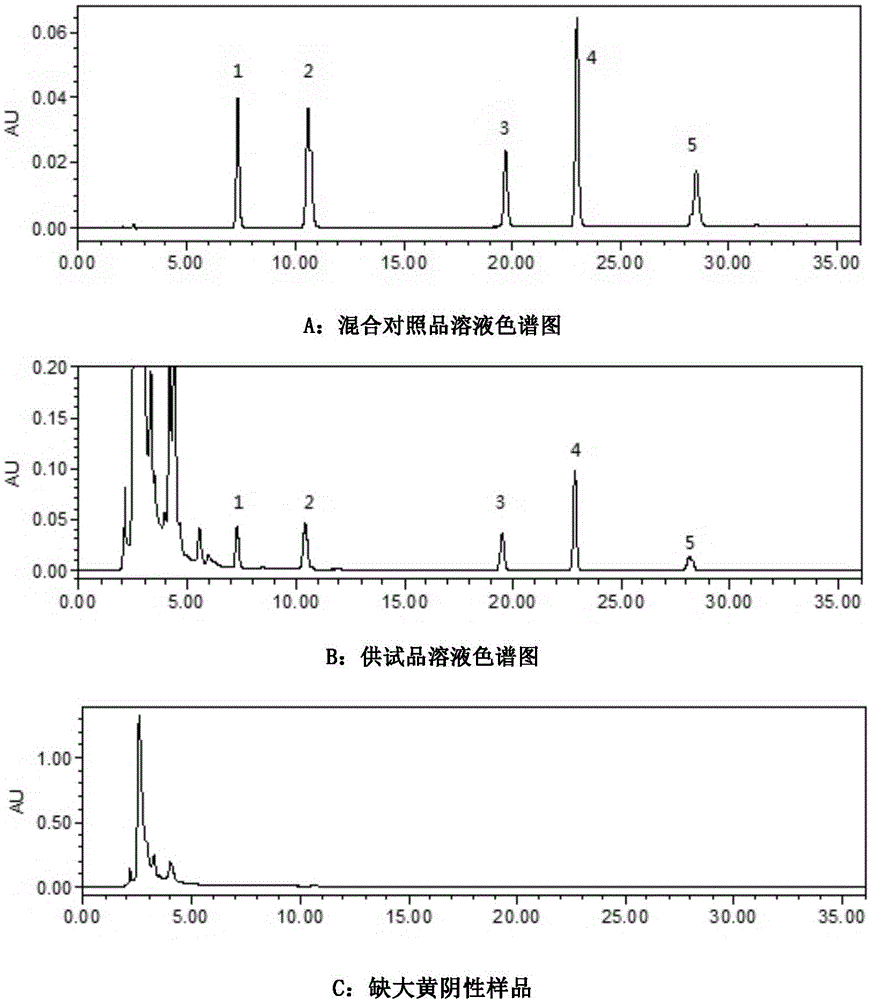
优选地,上述技术方案中,阴性对照溶液的制备:取缺大黄阴性样品,按游离蒽醌供试品溶液的制备方法制备成阴性样品溶液。
优选地,上述技术方案中,步骤(6)和(8)中的相对校正因子按照如下步骤确定:分别精密称取常温减压干燥12小时的芦荟大黄素对照品10.15mg、大黄酸对照品7.82mg、大黄素对照品6.83mg、大黄酚对照品8.66mg至100ml量瓶中,分别加甲醇溶解并定容至刻度,制成每1ml各含芦荟大黄素0.099267mg、大黄酸对照品0.0782mg、大黄素对照品0.0683mg、大黄酚对照品0.0866mg;精密称取大黄素甲醚对照品7.37mg至250ml量瓶中,加甲醇溶解并定容至刻度,制成每1ml含大黄素甲醚0.0288mg;按规定配制标准曲线溶液,分别吸取10μl注入高效液相色谱仪中,测定五个对照品的峰面积,按照如下公式分别计算芦荟大黄素、大黄酸、大黄酚及大黄素甲醚的相对校正因子,式中:c为质量浓度,s为峰面积。
优选地,上述技术方案中,本品每片含游离蒽醌以芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚的总量计,不得少于0.16mg。
优选地,上述技术方案中,本品每片含总蒽醌以芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚的总量计,不得少于1.2mg。
与现有技术相比,本发明具有如下有益效果:本发明在原标准hplc含量测定方法基础上,通过优化其提取方法和液相测定方法增订本品中五种总蒽醌及五种游离蒽醌的含量测定,该测定方法获得的检测数据能够同时对清火片中游离蒽醌和总蒽醌类成分的含量进行计算,该方法简便,分离和专属性较好,为清火片的质量控制提供了较为准确的参考,为清火片的质量控制标准的建立提供了依据。
附图说明:
图1为清火片中五种游离蒽醌成分含量测定hplc色谱图;
图2为清火片中五种总蒽醌成分含量测定hplc色谱图。
具体实施方式:
下面对本发明的具体实施方式进行详细描述,但应当理解本发明的保护范围并不受具体实施方式的限制。
除非另有其它明确表示,否则在整个说明书和权利要求书中,术语“包括”或其变换如“包含”或“包括有”等等将被理解为包括所陈述的组分或组成部分,而并未排除其它组分或其它组成部分。
一、游离蒽醌含量测定方法
色谱条件1
以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%-0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
色谱条件2
以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%-0.3%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
色谱条件3
以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.3%-0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
色谱条件4
以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.3%-0.5%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
对照品溶液的制备精密称取大黄素对照品适量,加甲醇制成每1ml含大黄素10μg的溶液,即得。
供试品溶液的制备取本品20片,除去包衣,精密称定,研细,取约0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,加热回流1小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,取续滤液,即得。余下续滤液备用。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,并按下式计算,即得。
游离蒽醌含量(mg/片)=(芦荟大黄素峰面积*0.716+大黄酸峰面积*0.874+大黄素峰面积*1.00+大黄酚峰面积*0.688+大黄素甲醚峰面积*1.007)*(大黄素对照品浓度/大黄素对照品峰面积)*稀释倍数*平均片重/称样量。
本品每片含游离蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于0.16mg。
二、总蒽醌含量测定方法
色谱条件与系统适用性试验同[含量测定]游离蒽醌项下。
对照品溶液的制备同[含量测定]游离蒽醌项下的对照品溶液,即得。
供试品溶液的制备取[含量测定]游离蒽醌项下的续滤液5ml,置烧瓶中,挥去甲醇,加8%盐酸溶液10ml,超声处理5分钟,再加三氯甲烷10ml,加热回流1小时,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10ml,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇适量使溶解,转移至10ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,并按游离蒽醌项下公式式计算,即得。
本品每片含总蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于1.2mg。
三、含量测定方法研究
(一)、游离蒽醌含量测定方法研究
1仪器与试药waterse2695高效液相色谱仪。超声波清洗器(型号:kq-300型,昆山市仪器有限公司)。色谱柱:thermoscientificc18,5um,4.6*250mm。
对照品及样品信息见表1、2。方法学验证采用清火片样品批号为:1312001。
表1样品信息
表2对照品信息
2提取方法选择
取样品研细后的粉末三份,取约0.4g,精密称定,置具塞锥形瓶中,分别精密加入甲醇25ml,分别加热回流30分钟,60分钟f90分钟,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,取续滤液,即得。测定五种上述成分总峰面积/w样。结果见表3。
表3甲醇回流时间的考察
结果表明,甲醇回流60分钟时,五种蒽醌总量较高,故采用甲醇回流60分钟的方法。
3检测波长的选择
参考原标准及中国药典2010年一部,检测波长为254nm。
4溶液的制备
对照品溶液的制备:同正文。
供试品溶液的制备:同成品正文。
阴性对照溶液的制备:取缺大黄阴性样品,按供试品溶液的制备方法制备成阴性样品溶液。
5样品含量测定结果
照正文含量测定方法测定两批样品中游离蒽醌含量,结果见表21。
表4二批样品含量测定结果
(二)、总蒽醌含量测定方法研究
1仪器与试药
同游离蒽醌含量测定项下。
2检测波长的选择
参考原标准及中国药典2010年一部,检测波长为254nm。
3溶液的制备
对照品溶液的制备:同正文。
供试品溶液的制备:同成品正文。
阴性对照溶液的制备:取缺大黄阴性样品,按供试品溶液的制备方法制备成阴性样品溶液。
4样品含量测定结果
照正文含量测定方法测定两批样品中总蒽醌含量,结果见表5。
表5二批样品含量测定结果
(三)本发明创造方法
1相对校正因子的确定
以大黄素对照品为参照,按照线性试验项下混合溶液的各成分峰面积,分别计算芦荟大黄素、大黄酸、大黄酚及大黄素甲醚的相对校正因子。芦荟大黄素、大黄酸、大黄酚及大黄素甲醚与内参物大黄酸间的相对校正因子分别为0.716、0.874、0.688、1.007,rsd分别为2.18%、2.09%、1.70%、3.39%,结果见表6。
表6相对校正因子表
其中:f=大黄素对照品浓度/大黄素对照品峰面积
蒽醌含量计算公式:
蒽醌含量(mg/g)=(芦荟大黄素峰面积*0.716+大黄酸峰面积*0.874+大黄素峰面积*1.00+大黄酚峰面积*0.688+大黄素甲醚峰面积*1.007)*(大黄素对照品浓度/大黄素对照品峰面积)*稀释倍数/称样量。
2外标法与本发明创造提供的检测方法含量测定结果的比较
采用外标法对清火片中五种蒽醌类成分进行含量测定,用建立的本发明的检测方法对其含量进行计算,将两种方法所得结果进行比较,以验证本发明的检测方法用于清火片蒽醌类成分含量测定的准确性。结果见表7-8。
表7采用两种方法对清火片中游离蒽醌总量的比较
表8采用两种方法对清火片中总蒽醌总量的比较
以大黄素为参照计算芦荟大黄素的相对校正因子
以大黄素为参照计算大黄酸的相对校正因子
以大黄素为参照计算大黄素甲醚的相对校正因子
(四)限度的确定
根据《中国药典》2010年版第一增补本中大黄含量的限度,含游离蒽醌不得少于0.20%,含总蒽醌不得少于1.5%。以及本品的处方和工艺,暂定本品每片含游离蒽醌,不得少于0.18mg。每片含总蒽醌,不得少于1.35mg。
(五)对照品外标法和本发明的检测方法实验方法
1、对照品外标法
【含量测定】游离蒽醌照高效液相色谱法(附录vid)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
对照品溶液的制备精密称取芦荟大黄素对照品、大黄酸对照品、大黄素对照品、大黄酚对照品、大黄素甲醚对照品适量,加甲醇分别制成每1ml含芦荟大黄素、大黄酸、大黄素、大黄酚各10μg,大黄素甲醚6μg的溶液,即得。
供试品溶液的制备取本品20片,除去包衣,精密称定,研细,取约0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,加热回流1小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,取续滤液,即得。余下续滤液备用。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,计算,即得。
本品每片含游离蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于0.18mg。
总蒽醌照高效液相色谱法(附录vid)测定。
色谱条件同[含量测定]游离蒽醌项下。
对照品溶液的制备同[含量测定]游离蒽醌项下的对照品溶液,即得。
供试品溶液的制备取[含量测定]游离蒽醌项下的续滤液5ml,置烧瓶中,挥去甲醇,加8%盐酸溶液10ml,超声处理5分钟,再加三氯甲烷10ml,加热回流1小时,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10ml,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇适量使溶解,转移至10ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,即得。
本品每片含总蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于1.35mg。
2、本发明的检测方法
【含量测定】游离蒽醌照高效液相色谱法(附录vid)测定。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以0.1%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;检测波长为254nm。理论板数按大黄素峰计算应不低于3000。
对照品溶液的制备精密称取大黄素对照品适量,加甲醇制成每1ml含大黄素10μg的溶液,即得。
供试品溶液的制备取本品20片,除去包衣,精密称定,研细,取约0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,加热回流1小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,取续滤液,即得。余下续滤液备用。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,并按下式计算,即得。
蒽醌含量(mg/片)=(芦荟大黄素峰面积*0.716+大黄酸峰面积*0.874+大黄素峰面积*1.00+大黄酚峰面积*0.688+大黄素甲醚峰面积*1.007)*(大黄素对照品浓度/大黄素对照品峰面积)*稀释倍数*平均片重/称样量
本品每片含游离蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于0.18mg。
总蒽醌照高效液相色谱法(附录vid)测定。
色谱条件与系统适用性试验同[含量测定]游离蒽醌项下。
对照品溶液的制备同[含量测定]游离蒽醌项下的对照品溶液,即得。
供试品溶液的制备取[含量测定]游离蒽醌项下的续滤液5ml,置烧瓶中,挥去甲醇,加8%盐酸溶液10ml,超声处理5分钟,再加三氯甲烷10ml,加热回流1小时,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10ml,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇适量使溶解,转移至10ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各10ul,注入液相色谱仪,测定,并按游离蒽醌项下公式式计算,即得。
本品每片含总蒽醌以芦荟大黄素(c15h10o5)、大黄酸(c15h8o6)、大黄素(c15h10o5)、大黄酚(c15h10o4)和大黄素甲醚(c16h12o5)的总量计,不得少于1.35mg。
前述对本发明的具体示例性实施方案的描述是为了说明和例证的目的。这些描述并非想将本发明限定为所公开的精确形式,并且很显然,根据上述教导,可以进行很多改变和变化。对示例性实施例进行选择和描述的目的在于解释本发明的特定原理及其实际应用,从而使得本领域的技术人员能够实现并利用本发明的各种不同的示例性实施方案以及各种不同的选择和改变。本发明的范围意在由权利要求书及其等同形式所限定。
- 还没有人留言评论。精彩留言会获得点赞!