一种采用HPLC-MS/MS测定食用香精香料中主要甜味剂的方法与流程
本发明属于食品添加剂理化指标检测技术领域,具体涉及一种采用hplc-ms/ms测定食用香精香料中主要甜味剂的方法。
背景技术:
食品及相关产品加工过程中经常用到香精、香料,随着社会对食品安全问题的关注,相关政府部门已经建立合理有效的监督管理和安全性评价法规制度,以规范香精、香料以及相关添加剂的生产经营和使用。甜味剂是一类重要的食品添加剂,用以改善食品口感和风味,其中非糖类甜味剂由于高甜度、低热量、多不参与代谢过程等优点而成为蔗糖的代用品在现代食品工业中被普遍应用。近年来,随着消费和认识水平的提高,人们对食品安全问题日益关注,食品中各种添加剂特别是甜味剂的使用情况越来越受到人们的重视。食品添加剂在规定的剂量范围内使用对人无害,若超量使用,则可能引起各种形式的毒性表现。因此,必须对其使用量进行严格管理,发挥其有利作用,防止其不利影响。目前,各国对糖精钠、甜蜜素等甜味剂采取了严格限制使用的政策,均制定了大量的标准,已成为国际贸易和国内标准中考察的重点。我国国家标准gb2760《食品添加剂使用标准》中对可用于食品加工的甜味剂,包括甜蜜素、安塞蜜、木糖醇、阿力甜、阿思巴甜等的适用范围和添加限量都有明确规定。
目前文献报道的有乳制品、白酒、饮料、包装纸张等食品中甜味剂的测定,也有香精中甜味剂的测定。甜味剂的测定方法主要有离子色谱法、毛细管电泳法、液相色谱法、液相色谱/串联质谱法等。由于人工合成甜味剂的物理化学性质、电化学性质、光谱性质的不同,液相色谱法很难满足多种甜味剂同时检测的要求,离子色谱法和毛细管电泳法也由于灵敏度低等原因应用不多。由于液相色谱/串联质谱的选择性强、灵敏度高以及抗基质干扰能力强,目前已广泛应用于食品添加剂的测定。但是,香精作为食品添加剂,其成分复杂、基质干扰严重,导致检测准确度较低。
技术实现要素:
为解决上述问题,本申请的采用hplc-ms/ms测定食用香精香料中主要甜味剂的方法,该方法采用基质溶剂配标,可以有效排除假阳性、提高了检测的准确性以及检测通量。
根据本申请的一个实施方案,
一种基于高效液相色谱-串联质谱法测定食用香精中主要甜味剂的方法,包括以下步骤:
(1)基质溶剂制备:以不含有目标物的食用香精为样品,以水为溶剂,超声萃取样品,萃取液过水相滤膜,制备基质溶剂;
(2)系列标准工作溶液制备:分别称取一定量的9种甜味剂标准品,以基质溶剂为配标溶剂,经逐级稀释,配置系列标准工作溶液;
(3)样品溶液制备:准确称取一定量的试样于离心管中,再准确移入一定体积的乙醇水溶液,用漩涡混合器分散均匀,再超声萃取,萃取液过水相滤膜,得样品进样液;
(4)高效液相色谱-串联质谱分析;用hplc-ms/ms色谱仪,对系列标准工作溶液和样品进样液分别进行检测;
(5)标准工作曲线建立及样品结果计算。
步骤(1)中,基质溶剂制备过程为:选取不含有木糖醇、安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、新橙皮苷、纽甜和甜菊糖等9种甜味剂的食用香精,准确称取0.5g样品,加10ml体积分数为10%的乙醇水溶液,涡旋混合使样品分散开,然后进行超声提取,于60%功率条件下超声萃取40min,结束后摇匀,静置片刻,经0.22µm水相滤膜过滤,得基质溶剂。
步骤(2)中,分别准确称取10mg木糖醇、10mg安赛蜜、10mg糖精钠、5mg甜蜜素、10mg三氯蔗糖、5mg阿斯巴甜、10mg新橙皮苷、5mg纽甜和5mg甜菊糖,于同一个100ml容量瓶中,精确至0.1mg,用基质溶剂溶解并定容,配制混合标准储备溶液。分别准确移取混合标准储备溶液50µl、100µl、250µl、0.5ml和1ml,用基质溶剂定容至10ml容量瓶中,混匀,配制得到5级标准工作溶液,各级系列溶液浓度如下,
系列标准工作溶液表
步骤(3)中,称取0.5g香精试样,精确至0.1mg,置于50ml具塞离心管中,准确移取1ml乙醇和9ml超纯水,涡旋混合使样品基质分散开,于60%功率条件下超声萃取40min,结束后摇匀,静置片刻,经0.22µm水相滤膜过滤,得样品进样液。
步骤(4)中,hplc-ms/ms分析,其中采用的高效液相色谱条件为:色谱柱:zorbaxeclipsexdb-c18,规格为150mm×4.6mm,填料粒度3.5µm;流动相a:甲醇,b:5mmol/l乙酸铵的水溶液,梯度洗脱见下表;柱温:40℃;进样量:5μl。
梯度洗脱程序表
步骤(4)中,hplc-ms/ms分析,其中采用的质谱条件为:离子源:电喷雾离子源(esi);离子源温度:100℃;干燥气温度:300℃;干燥气流量:9l/min;雾化气压力:40psi;毛细管电压:正离子为4000v,负离子为3500v;检测方式:多反应监测模式(mrm),参数如下,
目标物mrm参数条件表
步骤(5)中,以系列标准工作溶液中各目标物的浓度为横坐标,以色谱图中各目标物的峰面积为纵坐标,进行线性回归分析,得到各目标物的标准工作曲线。将相同条件下测得的样品进样液中各目标物的色谱峰面积,代入标准工作曲线,根据下列公式求得样品中各目标物的含量:
上式中:
x——试样中各目标物的含量,单位为毫克每千克(mg/kg);
c——由标准工作曲线上读取的样品进样液中各目标物的浓度,单位为毫克每升(µg/ml);
v——萃取体系的体积,单位为毫升(ml);
m——试样的质量,单位为克(g)。
与现有技术相比,本发明的基于高效液相色谱-串联质谱法测定食用香精中主要甜味剂的方法,具有如下优点:(1)本发明方法利用hplc-ms/ms同时测定食用香精中主要甜味剂的含量,抗基质干扰能力强,前处理方法简单;(2)本发明所用的萃取溶剂为10%体积分数的乙醇水溶液,对环境无污染,符合绿色发展理念;(3)本发明采用基质溶剂配置标准工作曲线,消除了质谱离子化过程中因基质效应给目标物定量产生的影响,能够准确同时检测食用香精中主要甜味剂含量(4)本发明具有较好的方法学考察结果,具有灵敏度高和重复性好的优点。
1、本方法的检测限:
将最低浓度的混合标准工作溶液进行hplc-ms/ms分析,以3倍信噪比(s/n=3)计算检测限(lod)。
2、本方法的加标回收率和重复性:
在一空白香精样品中加入甜味剂的标准溶液,然后分别进行前处理和hplc-ms/ms分析,并按照加标量和测定值计算其回收率。
各目标物的工作曲线(线性系数)、检测限、回收率和重复性如下表所示,
目标物的方法学考察
附图说明
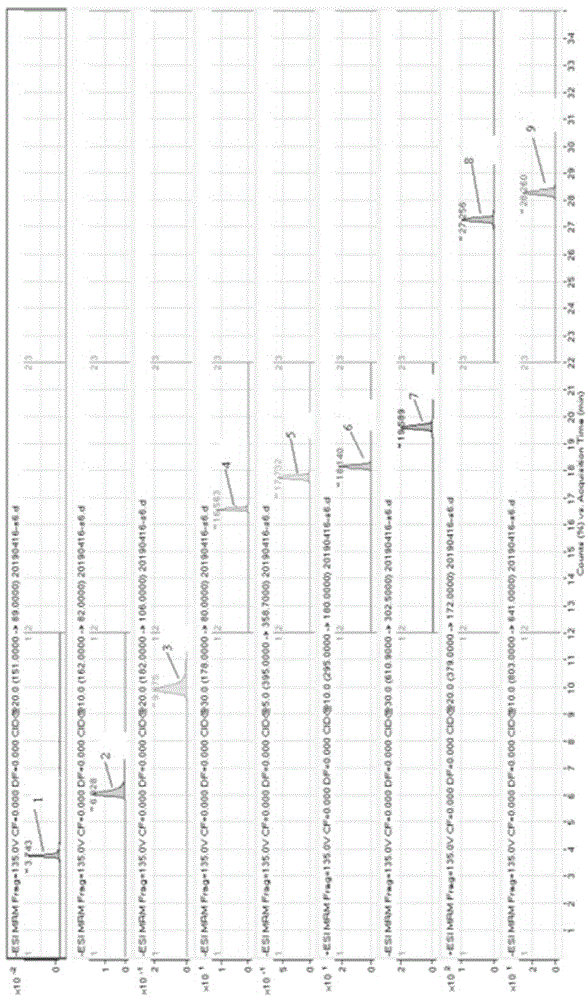
图1为实施例1标准工作溶液提取离子对色谱图。
图中,1为木糖醇(保留时间为3.74min),2为安赛蜜(保留时间为6.03min),3为糖精钠(保留时间9.88min),4为甜蜜素(保留时间16.56min),5为三氯蔗糖(保留时间17.73min),6为阿斯巴甜(保留时间18.14min),7为新橙皮苷(保留时间19.59min),8为纽甜(保留时间27.26min),9为甜菊糖(保留时间28.26min)。
具体实施方式
以下是本申请的具体实施例,对本申请的技术方案作进一步的描述,但本申请并不限于这些实施例。
实施例1
1、仪器与试剂
木糖醇、安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、新橙皮苷、纽甜
和甜菊糖等9种甜味剂的标准样品;甲醇、乙醇、乙酸铵(均为色谱纯);符合gb/t6682中一级水的要求。
6460hplc-ms/ms质谱仪(美国agilent公司),tm-1漩涡混合器(无锡
沃信仪器有限公司);kq-500de超声波发生器(昆山市超声仪器有限公司);电子天平(mettlertoledo公司,感量0.1mg);50ml离心管(corning公司,pp材质)。
2、基质溶剂制备
选取一不含有木糖醇、安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、
新橙皮苷、纽甜和甜菊糖等9种甜味剂的食用香精,准确称取0.5201g样品,加10ml体积分数为10%的乙醇水溶液,涡旋混合使样品分散开,然后进行超声提取,于60%功率条件下超声萃取40min,结束后摇匀,静置片刻,经0.22µm水相滤膜过滤,得基质溶剂。
3、系列标准工作溶液:配制方式见发明内容部分,此处不再重复。
4、样品处理
称取0.5105g香精试样1#,精确至0.1mg,置于50ml具塞离心管中,准确移取1ml乙醇和9ml超纯水,涡旋混合使样品基质分散开,于60%功率条件下超声萃取40min,结束后摇匀,静置片刻,经0.22µm水相滤膜过滤,得样品进样液。
5、hplc-ms/ms测定
6、结果计算
以外标法进行定量分析,即以各种甜味剂标准品的定量离子对峰面积对其相应浓度进行回归分析,得到各种甜味剂标准曲线,相关系数均大于等于0.995,对提取后的样品进行测定,得出目标物的定量离子对峰面积,代入标准曲线求得样品中甜味剂的含量,其中此样品中仅检出阿斯巴甜,含量为2.35mg/kg。
本文中所描述的具体实施例仅仅是对本申请精神作举例说明。本申请所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本申请的精神或者超越所附权利要求书所定义的范围。
- 还没有人留言评论。精彩留言会获得点赞!