一种定量检测水产品中吡咯素和甲基乙二醛氢咪唑酮的方法
本发明属于食品安全检测技术领域,具体涉及一种定量检测水产品中吡咯素和甲基乙二醛氢咪唑酮的方法。
背景技术:
食品加工实际上是食品各组分进行生物、物理和化学等一系列复杂的反应,尤其是富含高蛋白、高脂肪的食品在热处理过程中会伴随着美拉德反应的发生,导致美拉德反应产物——晚期糖化终末产物(ages)的形成。ages是由还原糖的羰基与氨基酸、蛋白质和含氮化合物的氨基之间的羰氨反应所形成。已有大量研究表明,过多的摄入膳食ages会导致体内ages的积累,从而造成糖尿病、肾病、阿尔兹海默症等多种慢性疾病的发生。已在食品中发现十余种ages,包括羧甲基赖氨酸、羧乙基赖氨酸、甲基乙二醛氢咪唑酮(mg-h1)、吡咯素(pyrraline)等。大多数研究主要聚焦于羧甲基赖氨酸和羧乙基赖氨酸这两种ages,目前,已发现吡咯素在油料制品中含量较高,而对于水产品中吡咯素检测方法的研究未见报道,且目前对于吡咯素的定量检测多采用外标法,没有添加内标对其含量进行校正,这造成吡咯素测定结果的不准确。水产品属于高蛋白高脂肪类食品,是ages的重要来源,因此定量检测水产品中ages对提升其食品安全水平具有很重要的现实意义。
食品体系复杂多样,对于食品中ages检测没有统一的标准,目前检测ages的方法主要是酶联免疫法和液质联用法。酶联免疫法对于抗体的要求较高,且受干扰较大;液相-质谱联用是较为广泛的使用方法,此方法对复杂样品具有高分离、高灵敏度、高选择性等优点,是检测食品中痕量物质的首选方法。然而,吡咯素极性较弱,适合用反向色谱柱对其进行分离和保留,而甲基乙二醛氢咪唑酮极性较强,目前大多数研究聚焦于羧甲基赖氨酸和羧乙基赖氨酸等高极性的ages,使用亲水性色谱柱可实现这类极性ages的同时保留和分离。所以当前所用的色谱柱很难做到对吡咯素和甲基乙二醛氢咪唑酮这两种ages的同时保留并分离,虽然可以通过使用九氟戊酸等离子对试剂增加保留效果,但是,离子对试剂会缩短色谱柱寿命、削弱液质电离效果,且会在质谱中残留、污染质谱。且不同的ages前处理方式不同,羧甲基赖氨酸和羧乙基赖氨酸等ages遇酸稳定,主要采用酸解法进行提取,吡咯素和甲基乙二醛氢咪唑酮遇酸不稳定,主要采用酶解法进行提取。
基于以上ages的性质和前处理方式的不同,对于吡咯素的研究大多为单独检测。因此,目前并没有同时定量检测水产品中极性较弱的吡咯素(pyrraline)和极性较强的甲基乙二醛氢咪唑酮(mg-h1)的方法。
技术实现要素:
针对现有技术中存在的问题,本发明目的在于提供一种定量检测水产品中吡咯素和甲基乙二醛氢咪唑酮的方法。
甲基乙二醛氢咪唑酮(mg-h1)的结构式如下:
吡咯素(pyrraline)的结构式如下:
本发明采用酶水解法提取吡咯素和甲基乙二醛氢咪唑酮,能够克服两者遇酸不稳定的缺点;利用固相萃取实现吡咯素和甲基乙二醛氢咪唑酮的除杂和富集,减少定量检测时的基质效应;采用内标法同时对吡咯素和甲基乙二醛氢咪唑酮进行定量,能够对两者含量进行校正;使用watersatlantist3(2.1×150mm,5μm)液相色谱柱,选用未添加离子对试剂的流动相,避免离子对试剂对质谱造成污染,然后利用液相-质谱联用对吡咯素和甲基乙二醛氢咪唑酮同时进行定量检测,实现在较短时间内同时更好地保留并定量检测水产品中极性较弱的吡咯素和极性较强的甲基乙二醛氢咪唑酮,提高了检测效率,且检测方法灵敏度高。
为了达到上述目的,本发明采用如下技术方案:
一种定量检测水产品中吡咯素和甲基乙二醛氢咪唑酮的方法,步骤如下:
步骤1:样品中吡咯素和甲基乙二醛氢咪唑酮的提取
1.1采用酶解法提取样品中吡咯素和甲基乙二醛氢咪唑酮;
1.2向酶解后的样品溶液中加入内标用以校正误差,并过滤膜除去杂质;
1.3加标后的样品溶液过固相萃取柱进行萃取,收集洗脱液;
步骤2:将步骤1获得的洗脱液利用液相-质谱联用进行定量检测;
步骤3:绘制标准曲线:向标准品中加入内标后采用液相-质谱联用进行检测,以标准品浓度与内标浓度比为横坐标,标准品峰面积与内标峰面积比为纵坐标绘制标准曲线,得到方程;
步骤4:将步骤2获得的检测数据对照步骤3的标准曲线和方程,计算获得样品中吡咯素和甲基乙二醛氢咪唑酮的浓度。
在上述方案的基础上,所述酶解法提取样品中吡咯素和甲基乙二醛氢咪唑酮具体操作为:
①称取20mg样品于1ml20mmhcl溶液中,分别加入40μl6mg/ml胃蛋白酶溶液和20μl2mg/ml百里香酚溶液,充入氮气,迅速封口,然后37℃震摇24h;
②用260mmkoh溶液将步骤①反应后的体系ph调至7.4,加入300μl0.5m磷酸钾缓冲液和1ml2mg/ml链酶蛋白酶(xiv型)溶液,充入氮气,迅速封口,然后37℃震摇24h;
③向步骤②反应后的体系中依次加入100μl亮氨酸氨基肽酶m(vi-s型)溶液和50μl脯氨酸肽酶溶液,然后充入氮气,迅速封口,37℃震摇24h。
所述胃蛋白酶溶液的溶剂为20mmhcl溶液;酶活力为2400u/ml;
所述百里香酚溶液的溶剂为20mmhcl溶液;
所述链酶蛋白酶(xiv型)溶液的溶剂为10mmpbs缓冲液;酶活力为7u/ml;
所述亮氨酸氨基肽酶m(vi-s型)溶液的溶剂为10mmpbs缓冲液;酶活力6u/样品;
所述脯氨酸肽酶溶液溶剂为10mmpbs缓冲液;酶活力为8u/样品。
本发明中百里香酚具有广谱抗菌性和防腐功能,在体系中加入百里香酚溶液能够抑制多种微生物的生长繁殖;加入磷酸钾缓冲溶液使体系ph在一个适当的阈值内,保证酶溶液发挥其活性。
在上述方案的基础上,所述步骤1中采用的内标为mg-h1-d3。
在上述方案的基础上,所述固相萃取柱萃取方法为:
取3ml甲醇活化hlbpro萃取柱,然后取3ml超纯水平衡hlbpro萃取柱,取1ml步骤1.2加标后的酶解样品溶液,过hlbpro萃取柱,取3ml超纯水除杂,然后用3ml5%氨水甲醇溶液洗脱,收集洗脱液于60℃氮吹干燥,最后复溶于10%乙腈水溶液。
在上述方案的基础上,所述固相萃取柱为cnwpoly-seryhlbpro,规格为60mg/3ml。
本发明采用的hlbpro萃取柱具备极端耐酸碱性的特点,流速较快,过柱时间短,同时适用于极性、非极性的化合物。
在上述方案的基础上,所述液相-质谱联用的液相色谱条件为:
色谱柱:watersatlantist3(2.1×150mm,5μm);流动相:含0.1%甲酸和10%乙腈的水溶液;流速:0.2ml/min;柱温:30℃;进样体积:10μl;梯度洗脱程序:等梯度洗脱。
在上述方案的基础上,所述液相-质谱联用的质谱条件为:
电喷雾离子源(esi);正离子扫描模式;多反应监测模式(mrm);干燥气温温度为350℃;干燥气流速为12l/min;雾化器压力为45psi;毛细管电压为3500v;碎裂电压为135v。
在上述方案的基础上,液相-质谱联用中吡咯素母离子质荷比为255,定量检测特征碎片离子质荷比为175;定性检测特征碎片离子质荷比为237;甲基乙二醛氢咪唑酮母离子质荷比为229,定量检测特征碎片离子质荷比为70,定性检测特征碎片离子质荷比为114。
在上述方案的基础上,所述标准曲线绘制中标准溶液的浓度范围为0.01-1μg/ml。
在上述方案的基础上,所述水产品为鲟鱼。
本发明技术方案的优点:
1.本发明采用酶水解法同时提取水产品中吡咯素和甲基乙二醛氢咪唑酮,能够克服两者遇酸不稳定的缺点;
2.采用内标法同时对吡咯素和甲基乙二醛氢咪唑酮进行定量,能够对两者含量进行校正;
3.本发明可在较短时间内同时更好地保留并定量检测极性较弱的吡咯素和极性较强的甲基乙二醛氢咪唑酮,提高了检测效率;
4.本发明具有较好的分离效果,灵敏度高,且线性关系好,r2均在0.998以上;
5.本发明实现水产品中吡咯素与甲基乙二醛氢咪唑酮的同时检测。
6.本发明使用未添加离子对试剂的流动相,避免造成质谱的污染。
附图说明
图1为吡咯素标准曲线;
图2为甲基乙二醛氢咪唑酮标准曲线;
图3为标准品溶液的总离子色谱图;
图4为样品目标化合物质谱图;
图5为甲基乙二醛氢咪唑酮和吡咯素的总离子色谱图;
图6为甲基乙二醛氢咪唑酮和吡咯素的总离子色谱图;
图7为甲基乙二醛氢咪唑酮和吡咯素的总离子色谱图。
具体实施方式
在本发明中所使用的术语,除非有另外说明,一般具有本领域普通技术人员通常理解的含义。
下面结合具体实施例,并参照数据进一步详细的描述本发明。以下实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。
实施例1
1.试剂和设备
(1)试剂
吡咯素标准品加拿大torontoresearchchemicals
甲基乙二醛氢咪唑酮标准品美国internationallaboratory
甲基乙二醛氢咪唑酮内标标准品美国bioberry
胃蛋白酶、链蛋白酶(xiv型)、亮氨酸氨基肽酶m(vi-s型)、脯氨酸肽酶sigma-aldrich
磷酸二氢钾、氢氧化钾、pbs、盐酸、甲醇、乙腈、氨水国药集团化学试剂有限公司
(2)设备
6410triplequad质谱仪美国agilenttechnologies
1260infinity液相色谱仪美国agilenttechnologies
分析天平北京赛多利斯科学仪器有限公司
xw-80a微型涡旋混合仪上海沪西分析仪器厂有限公司
kq-300vde型双频数控超声波清洗器昆山市超声仪器有限公司
n-20多功能氮吹仪山东云网数据科技有限公司
cnwpoly-seryhlbpro固相萃取柱上海安谱实验科技股份有限公司
lc-cq-24f固相萃取装置上海力辰邦西仪器科技有限公司
2.提取吡咯素和甲基乙二醛氢咪唑酮
2.1称取蒸煮20min冻干的鲟鱼样品20mg于1ml20mmhcl溶液中,分别加入40μl6mg/ml胃蛋白酶溶液(酶活力为2400u/ml)和20μl2mg/ml百里香酚溶液(两者溶剂均为20mmhcl),充入氮气,迅速盖帽,然后37℃震摇24h;
2.2用260mmkoh溶液将上述体系ph调至7.4,加入300μl0.5m磷酸钾缓冲液和1ml2mg/ml链酶蛋白酶(xiv型)溶液(溶剂为10mmpbs缓冲液,酶活力为7u/ml),充入氮气,迅速盖帽,然后37℃震摇24h;
2.3向上述体系中分别加入100μl亮氨酸氨基肽酶m(vi-s型)溶液(使每个样品酶活为6u,溶剂为10mmpbs缓冲液)和50μl脯氨酸肽酶溶液(使每个样品酶活为8u,溶剂为10mmpbs缓冲液),然后充入氮气,迅速盖帽,37℃震摇24h;
3.固相萃取
取3ml甲醇活化hlbpro萃取柱(cnwpoly-seryhlbpro,规格60mg/3ml),然后取3ml超纯水平衡hlbpro萃取柱,取1ml步骤2.3酶解液,向其中加入20μl5μg/ml甲基乙二醛氢咪唑酮内标(mg-h1-d3)过0.22μm滤膜,然后过hlbpro萃取柱,取3ml超纯水除杂,真空抽干后用3ml5%氨水甲醇溶液洗脱,收集洗脱液于60℃氮吹干燥,最后复溶于10%乙腈水溶液。
4.配置混合标准溶液(如表1),用浓度均为5μg/ml的mg-h1-d3、mg-h1、pyrraline标准品配置成1ml含0.1μg/ml内标的0、0.01、0.05、0.1、0.2、0.4、0.6、0.8、1μg/ml的标准溶液。
混合标准溶液浓度范围为0.01μg~1μg/ml,将混合标准溶液和上述固相萃取液采用液相-质谱联用进行定量检测。
表1标准溶液的配制方法
5.液相-质谱联用检测
5.1液相色谱条件
色谱柱:watersatlantist3(2.1×150mm,5μm);
流动相:含0.1%甲酸和10%乙腈的水溶液;
流速:0.2ml/min;
柱温:30℃;
进样体积:10μl;
梯度洗脱程序:等梯度洗脱。
5.2质谱条件
电喷雾离子源(esi);
正离子扫描模式;
多反应监测模式(mrm);
干燥气温温度为350℃;
干燥气流速为12l/min;
雾化器压力为45psi;
毛细管电压为3500v;
碎裂电压为135v。
5.3制作标准曲线
横坐标为吡咯素浓度(或甲基乙二醛氢咪唑酮浓度)与甲基乙二醛氢咪唑酮内标浓度比,纵坐标为吡咯素峰面积(或甲基乙二醛氢咪唑酮峰面积)与甲基乙二醛氢咪唑酮峰内标面积比。得到吡咯素标准曲线为y=1.8021x+0.3551,r2=0.9985(如图1);甲基乙二醛氢咪唑酮标准曲线为y=0.5726x-0.0029,r2=0.9987(如图2),其中x为浓度比值,y为峰面积比值。图3为标准品溶液的总离子色谱图,甲基乙二醛氢咪唑酮先出峰,出峰时间为1.91min;吡咯素后出峰,出峰时间为4.8min。证明该色谱条件分离效果较好,能够把这两种ages分开。
5.4将上述酶解液采用液相-质谱联用进行定量检测,如图4为样品目标化合物的质谱图,吡咯素母离子质荷比为255,定量检测特征碎片离子质荷比为175;定性检测特征碎片离子质荷比为237;甲基乙二醛氢咪唑酮母离子质荷比为229,定量检测特征碎片离子质荷比为70,定性检测特征碎片离子质荷比为114。利用多反应检测法获取质谱数据如图4,可以看出图中出现了质荷比为70、114和175、237的质谱图,分别对应甲基乙二醛氢咪唑酮和吡咯素的特征碎片离子,证明该质谱条件可以同时检测出吡咯素和甲基乙二醛氢咪唑酮这两种ages。且实验结果表明当吡咯素和甲基乙二醛氢咪唑酮的浓度为0.01μg/ml时,信噪比(s/n)均大于10,说明该方法的定量限(loq)可以达到0.01μg/ml。
对比例1
将实施例1步骤3中的hlbpro萃取柱替换为poly-sery-mcx固相萃取柱,其它条件不变。
检测结果发现,hlbpro萃取柱被替换后,吡咯素的萃取效果较差。
对比例2
将实施例1步骤5.1中的色谱柱替换为waterssymmetryc18(2.1×100mm,3.5μm),其它条件不变。
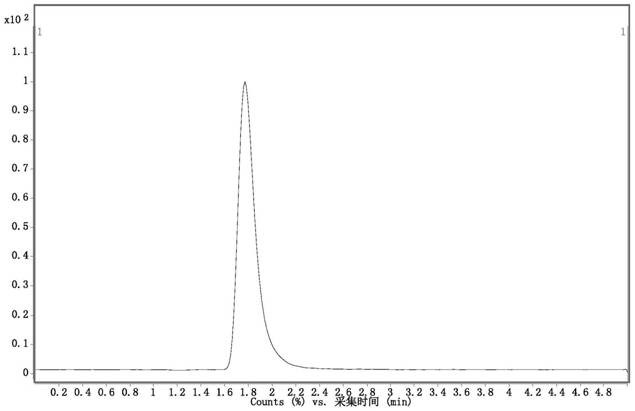
检测结果如图5所示,吡咯素在1.77min出峰,甲基乙二醛氢咪唑酮未出峰,说明甲基乙二醛氢咪唑酮未在色谱柱上保留。
对比例3
将实施例1步骤5.1中的色谱柱替换为agilentzorbaxsb-c18(4.6×150mm),其它条件不变。
检测结果如图6所示,吡咯素和甲基乙二醛氢咪唑酮均未出峰,证明两者在色谱柱上均未保留。
对比例4
将实施例1步骤5.1中的流动相替换为含0.1%甲酸和5%乙腈的水溶液,其它条件不变。
检测结果发现,当流动相为含0.1%甲酸和10%乙腈的水溶液时,甲基乙二醛氢咪唑酮在1.91min出峰,吡咯素在4.8min出峰。当流动相为含0.1%甲酸和5%乙腈的水溶液时,如图7所示,甲基乙二醛氢咪唑酮出峰时间不变,而吡咯素出峰时间在5.13min,增加了时间成本。
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!