基因组IIB型限制内切酶位点预测方法及电子设备与流程
本发明涉及基因相关技术领域,特别是一种基因组iib型限制内切酶位点预测方法及电子设备。
背景技术:
iib型限制内切酶不同于传统限制内切酶,其能够识别特定序列并切断与识别位点相距一定距离的两翼序列(图1)。目前iib型限制内切酶共报道有27种,其中11种能够在试剂公司购买到。
由于iib型限制内切酶能够识别dna特异位点并切割固定长度的特性,近几年开发出一种利用iib型限制内切酶与radseq高通量测序技术相结合的方法(2b-rad)。其实验过程首先通过iib型限制内切酶对基因组dna进行切割,回收等长酶切短序列,利用切割产生的粘性末端与测序接头连接后通过pcr进行扩增产生高通量测序文库,最后利用高通量测序仪对文库进行测序后,对测序结果进行分析。利用2b-rad的可以构建基因组遗传图谱、动物性别鉴定、种群遗传学研究、种间亲缘关系研究、遗传育种研究、基因组遗传突变研究、微生物分型等。由此可见,2b-rad在科研研究中应用的多样性与广泛。
由于iib型限制内切酶在基因组中是随机分布,不同物种基因组大小不同影响着iib型限制内切酶的识别位点数量,不同iib型限制内切酶对同一个基因组的酶切位点识别数量也各不相同,并且由于iib型限制内切酶能够切割基因组的长度有限,不能保证酶切片段序列的特异性。在2b-rad在研究与应用中的应用中,需要对基因组进行iib型限制内切酶酶切位点进行预测并且统计酶切位点数量、比较酶切片段序列特异性以及计算酶切位点跨度的均匀性,以此来预选最佳iib型限制内切酶种类,选择合适高通量测序深度,选择最优测序文库接头等。
然而,现有的iib型限制内切酶切割基因组dna技术中,仅依靠预实验进行iib型限制内切酶对dna的切割,来实现对酶切位点的统计,导致浪费过多的时间成本与经费。
技术实现要素:
基于此,有必要针对现有技术仅依靠预实验进行iib型限制内切酶对dna的切割,来实现对酶切位点的统计的技术问题,提供一种基因组iib型限制内切酶位点预测方法及电子设备。
本发明提供一种基因组iib型限制内切酶位点预测方法,包括:
获取待预测iib型限制内切酶、以及待预测基因组信息;
对所述待预测基因组内基于所有所述待预测iib型限制内切酶进行酶切位点预测,得到所有酶切位置信息;
根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列;
根据所述酶切位置信息,计算并输出酶切片段序列之间的间隔长度;
对所述间隔长度进行统计。
进一步的,所述根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列之后,还包括:
获取所述待预测iib型限制内切酶对应的酶切片段序列正则表达式,根据所述酶切片段序列与所述酶切片段序列正则表达式的匹配结果,判断所述酶切片段序列为正向序列或反向序列。
更进一步的,所述判断所述酶切片段序列为正向序列或反向序列之后,还包括:
将判断为反向序列的酶切片段序列进行反向互补处理。
进一步的,所述统计并输出所述待预测基因组中所有所述iib型限制内切酶酶切片段序列之后,还包括:
对所有所述酶切片段序列进行比较,统计并输出所有所述酶切片段序列中的单一酶切片段序列、重复酶切片段序列、以及重复酶切片段序列的重复数量。
进一步的,所述对所述间隔长度进行统计之后,还包括:
所述间隔长度的统计结果,生成用于表示每个所述间隔长度的数量的统计堆积图。
本发明提供一种电子设备,包括:
至少一个处理器;以及,
与所述至少一个处理器通信连接的存储器;其中,
所述存储器存储有可被所述一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够:
获取待预测iib型限制内切酶、以及待预测基因组信息;
对所述待预测基因组基于所述待预测iib型限制内切酶进行酶切位点预测,得到所有酶切位置信息;
根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列;
根据所述酶切位置信息,计算并输出酶切片段序列之间的间隔长度;
对所述间隔长度进行统计。
进一步的,所述根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列之后,还包括:
获取所述待预测iib型限制内切酶对应的酶切片段序列正则表达式,根据所述酶切片段序列与所述酶切片段序列正则表达式的匹配结果,判断所述酶切片段序列为正向序列或反向序列。
更进一步的,所述判断所述酶切片段序列为正向序列或反向序列之后,还包括:
将判断为反向序列的酶切片段序列进行反向互补处理。
进一步的,所述统计并输出所述待预测基因组中所有所述iib型限制内切酶酶切片段序列之后,还包括:
对所有所述酶切片段序列进行比较,统计并输出所有所述酶切片段序列中的单一酶切片段序列、重复酶切片段序列、以及重复酶切片段序列的重复次数。
进一步的,所述对所述间隔长度进行统计之后,还包括:
所述间隔长度的统计结果,生成用于表示每个所述间隔长度的数量的统计堆积图。
本发明通过计算机模拟iib型限制内切酶对dna的切割并对酶切位点进行统计,为后续实验做参考,避免在预实验上浪费过多的时间成本与经费。
附图说明
图1为本发明一种基因组iib型限制内切酶位点预测方法的工作流程图;
图2为本发明最佳实施例的工作流程图;
图3为本发明一种电子设备的硬件结构示意图。
具体实施方式
下面结合附图和具体实施例对本发明做进一步详细的说明。
如图1所示为本发明一种基因组iib型限制内切酶位点预测方法的工作流程图,包括:
步骤s101,获取待预测iib型限制内切酶、以及待预测基因组信息;
步骤s102,对所述待预测基因组基于所有所述待预测iib型限制内切酶进行酶切位点预测,得到所有酶切位置信息;
步骤s103,根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列;
步骤s104,根据所述酶切位置信息,计算并输出酶切片段序列之间的间隔长度;
步骤s105,对所述间隔长度进行统计。
具体来说,可以使用python编程语言执行步骤s101~步骤s105,对dna序列内拥有的iib型限制内切酶酶切位点进行预测。步骤s101可以获取待预测iib型限制内切酶的种类名来确定对应的待预测iib型限制内切酶、通过输入待预测基因组的序列来确定待预测基因组,在步骤s102中,可以使用例如python的biopython模块进行酶切位点预测,得到预测的酶切位置后,在步骤s103中,根据酶切位置确定酶切片段序列并输出,酶切片段序列可以输出为fasta序列格式,在步骤s104中计算酶切位置的间隔长度,然后在步骤s105中对间隔长度进行统计。
本发明通过计算机模拟iib型限制内切酶对dna的切割并对酶切位点进行统计,为后续实验做参考,避免在预实验上浪费过多的时间成本与经费。
在其中一个实施例中,所述根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列之后,还包括:
获取所述待预测iib型限制内切酶对应的酶切片段序列正则表达式,根据所述酶切片段序列与所述酶切片段序列正则表达式的匹配结果,判断所述酶切片段序列为正向序列或反向序列。
具体来说,通过在步骤s101中获取的待预测iib型限制内切酶,计算其酶切长度与粘性切口长度,从而建立识别其酶切片段序列的正则表达式。正则表达式可以为多个,每个正则表达式表示一种酶切片段序列的特征,例如:(‘r’,[ag])表示ra或者rg的酶切片段序列。由于基因组输入时可以为正向也可以为反向,因此,采用酶切位置计算酶切片段序列所得出的酶切片段序列可能为正向也可能为反向。本实施例通过酶切片段序列正则表达式来匹配酶切片段序列,从而判断酶切片段序列为正向或者反向。
在其中一个实施例中,所述判断所述酶切片段序列为正向序列或反向序列之后,还包括:
将判断为反向序列的酶切片段序列进行反向互补处理。
本实施例将判断为反向序列的酶切片段序列进行反向互补处理,从而避免正反向酶切片段序列被错误区分为不同序列。
在其中一个实施例中,所述统计并输出所述待预测基因组中所有所述iib型限制内切酶酶切片段序列之后,还包括:
对所有所述酶切片段序列进行比较,统计并输出所有所述酶切片段序列中的单一酶切片段序列、重复酶切片段序列、以及重复酶切片段序列的重复次数。
本实施例统计酶切片段序列中的单一酶切片段序列和重复酶切片段序列与重复次数,便于后续进行高通测序。
在其中一个实施例中,所述对所述间隔长度进行统计之后,还包括:
所述间隔长度的统计结果,生成用于表示每个所述间隔长度的数量的统计堆积图。
本实施例通过统计堆积图,以直观的方法显示酶切结果。
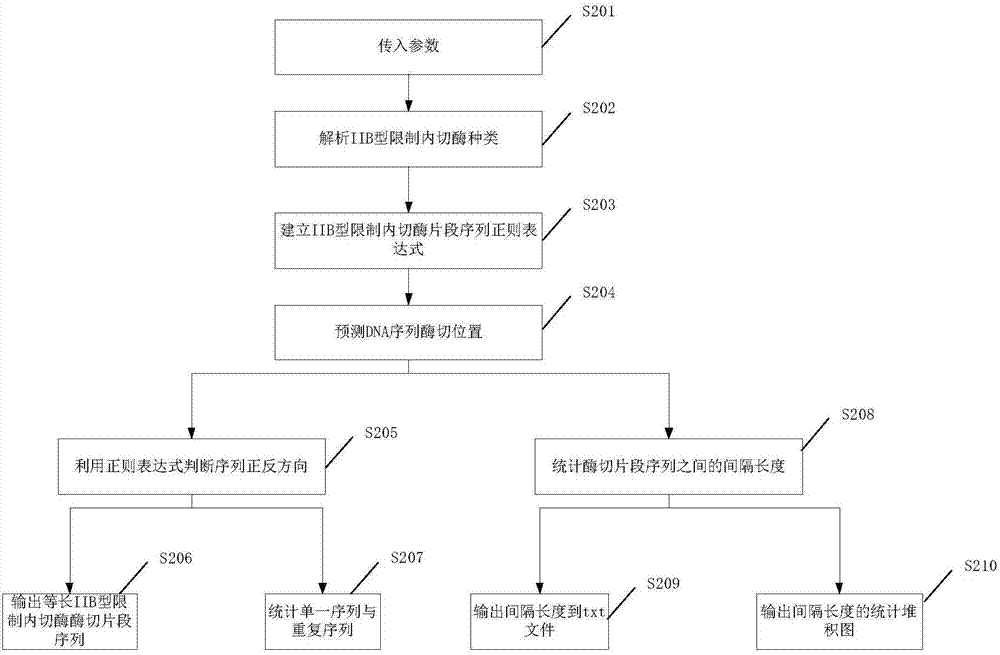
如图2所示为本发明最佳实施例的工作流程图,该方法是用python语言编程,导入的模块包括sys、getopt、re、biopython、matplotlib和skimage,方法包括:
步骤s201,利用sys、getopt两个模块传入三个参数包括输入文件名、输出文件名和iib型限制内切酶的种类名。
步骤s202,通过传入的iib型限制内切酶的种类名,确定iib型限制内切酶;
步骤s203,计算iib型限制内切酶的酶切长度与粘性切口长度,建立识别其酶切片段序列的正则表达式;
步骤s204,利用biopython模块对输入文件名所指定的基因组基于iib型限制内切酶进行酶切位点预测得到多个酶切位置,基于酶切位置确定酶切片段序列,分别执行步骤s205和步骤s208;
步骤s205,酶切片段序列经过正则表达式匹配、判断正反向;
步骤s206,输出fasta格式的序列到一个文件中,fasta格式的序列名为dna序列加位置和正反向信息;
步骤s207,通过比较所有酶切片段序列,计算特有的单一片段的数量与重复片段的数量,输出到显示屏;
步骤s208,统计酶切片段序列之间的间隔长度;
步骤s209,把间隔长度输出到一个txt文件中;
步骤s210,利用matplotlib和skimage两个模块输出间隔长度的堆积图,以直观的方法显示酶切结果。
如图3所示为本发明一种电子设备的硬件结构示意图,包括:
至少一个处理器301;以及,
与所述至少一个处理器301通信连接的存储器302;其中,
所述存储器302存储有可被所述一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够:
获取待预测iib型限制内切酶、以及待预测基因组信息;
对所述待预测基因组内基于所有所述待预测iib型限制内切酶进行酶切位点预测,得到所有酶切位置信息;
根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列;
根据所述酶切位置信息,计算并输出酶切片段序列之间的间隔长度;
对所述间隔长度进行统计。
图3中以一个处理器302为例。
电子设备还可以包括:输入装置303和输出装置304。
处理器301、存储器302、输入装置303及显示装置304可以通过总线或者其他方式连接,图中以通过总线连接为例。
存储器302作为一种非易失性计算机可读存储介质,可用于存储非易失性软件程序、非易失性计算机可执行程序以及模块,如本申请实施例中的基因组iib型限制内切酶位点预测方法对应的程序指令/模块,例如,图1、图2所示的方法流程。处理器301通过运行存储在存储器302中的非易失性软件程序、指令以及模块,从而执行各种功能应用以及数据处理,即实现上述实施例中的基因组iib型限制内切酶位点预测方法。
存储器302可以包括存储程序区和存储数据区,其中,存储程序区可存储操作系统、至少一个功能所需要的应用程序;存储数据区可存储根据基因组iib型限制内切酶位点预测方法的使用所创建的数据等。此外,存储器302可以包括高速随机存取存储器,还可以包括非易失性存储器,例如至少一个磁盘存储器件、闪存器件、或其他非易失性固态存储器件。在一些实施例中,存储器302可选包括相对于处理器301远程设置的存储器,这些远程存储器可以通过网络连接至执行基因组iib型限制内切酶位点预测方法的装置。上述网络的实例包括但不限于互联网、企业内部网、局域网、移动通信网及其组合。
输入装置303可接收输入的用户点击,以及产生与基因组iib型限制内切酶位点预测方法的用户设置以及功能控制有关的信号输入。显示装置304可包括显示屏等显示设备。
在所述一个或者多个模块存储在所述存储器302中,当被所述一个或者多个处理器301运行时,执行上述任意方法实施例中的基因组iib型限制内切酶位点预测方法。
在其中一个实施例中,所述根据所述酶切位置信息,统计并输出所述待预测基因组中所有所述待预测iib型限制内切酶对应的酶切片段序列之后,还包括:
获取所述待预测iib型限制内切酶对应的酶切片段序列正则表达式,根据所述酶切片段序列与所述酶切片段序列正则表达式的匹配结果,判断所述酶切片段序列为正向序列或反向序列。
在其中一个实施例中,所述判断所述酶切片段序列为正向序列或反向序列之后,还包括:
将判断为反向序列的酶切片段序列进行反向互补处理。
在其中一个实施例中,所述统计并输出所述待预测基因组中所有所述iib型限制内切酶酶切片段序列之后,还包括:
对所有所述酶切片段序列进行比较,统计并输出所有所述酶切片段序列中的单一酶切片段序列、重复酶切片段序列、以及重复酶切片段序列重复次数。
在其中一个实施例中,所述对所述间隔长度进行统计之后,还包括:
所述间隔长度的统计结果,生成用于表示每个所述间隔长度的数量的统计堆积图。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!