含氢过渡金属氧化物、制备方法及原电池与流程
本发明涉及材料技术领域,尤其涉及一种含氢过渡金属氧化物、制备方法及原电池。
背景技术:
传统对氧化物氢化的方法都是通过热学的方法实现的,如通过一些氢化物(cah2,nah)对氧化物进行还原。这些h离子会替代氧化物中的o,形成h-m键(m:过渡金属),由于h-m键比m-o短,因此这些氢化的氧化物的特征表现为晶格体积的变小。过渡金属氧化物的氢化,一是可以改变其结构相;二是会伴随着电子或者空穴的掺杂,改变材料的电学或磁学特性。此外,氢化的过程中有时也会将氧化物中的氧带走,变成一些缺氧的相。通过氢热还原的方法,一些含氢的氧化物已经被制备出来,如氢化的lasrcoo3、batio3、vo2、tio2等。对于实现材料结构的结构相变,除了氢化的方法,还可以通过热氧化的方法实现。如通过高氧压氧化的方法,可以实现钙铁石结构srcoo2.5到钙钛矿结构srcoo3的转变。
上述这些方法都局限于两相之间的调控。传统方案中,并没有一种能够实现三相变化的含氢过渡金属氧化物以及电场调控该含氢过渡金属氧化物实现三相相变的方法。
技术实现要素:
基于此,有必要针对上述技术问题,提供一种含氢过渡金属氧化物、制备方法及原电池,所述含氢过渡金属氧化物能够实现三相变化。
一种含氢过渡金属氧化物,该含氢过渡金属氧化物的结构式为aboxhy,其中a为碱土金属元素和稀土金属元素中的一种或多种,b为过渡族金属元素中的一种或多种,x的取值范围为1-3,y的取值范围为0-2.5。
在其中一个实施例中,碱土金属元素包括be、mg、ca、sr和ba,所述稀土金属元素包括la、ce、pr、nd、pm、sm、eu、gd、tb、dy、ho、er、tm和yb,所述过渡族金属元素包括co、cr、fe、mn、ni、cu、ti、zn、sc和v。
在其中一个实施例中,b为过渡族金属元素co。
在其中一个实施例中,a为碱土金属元素sr。
在其中一个实施例中,x为2.5,y为0-2.5。
一种含氢过渡金属氧化物的制备方法,包括以下步骤:
s100,提供一种结构式为aboz的过渡金属氧化物,其中,z大于等于2且小于等于3;
s200,将所述过渡金属氧化物浸入离子液体中,所述离子液体中包含氢离子和氧离子;
s300,对所述过渡金属氧化物施加电场,使离子液体中的氢离子插入所述过渡金属氧化物中。
在其中一个实施例中,所述步骤s100包括以下步骤:
s110,提供基底;
s120,在所述基底表面沉积形成结构式为aboz的过渡金属氧化物薄膜;
s130,在所述过渡金属氧化物薄膜的表面形成第一电极。
在其中一个实施例中,所述基底为陶瓷基底、硅基底、玻璃基底、金属基底或者聚合物中的一种,所述步骤s120通过脉冲激光沉积的方法在所述基底表面外延生长获得所述过渡金属氧化物薄膜。
在其中一个实施例中,所述步骤s130中,所述第一电极接触过渡金属氧化物薄膜,形成底电极。
在其中一个实施例中,所述步骤s300包括以下步骤:
s310,提供第二电极以及电源;
s320,将所述第二电极与所述第一电极间隔设置并分别与所述电源电连接;
s330,将所述第二电极浸入所述离子液体中,并通过所述电源施加从所述第二电极到所述第一电极方向的电场。
一种原电池,包括阴极电极、与所述阴极电极间隔设置的阳极电极,以及设置与所述阴极电极与所述阳极电极之间的电解质,其特征在于,所述阴极电极和所述阳极电极为上述实施例中所述的含氢过渡金属氧化物。
本发明通过电场控制氢化的方法实现了一种具有新晶体结构的含氢过渡金属氧化物以及制备方法,结合电场控制氢化和氧化的方法,所述含氢过渡金属氧化物实现了三个不同结构相之间在电场下的可控转变,并实现了丰富的物态和物性调控。另外,本发明还提供了一种利用上述含氢过渡金属氧化物做为电极的原电池。
附图说明
图1为本发明实施例提供的含氢过渡金属氧化物的制备方法的流程图;
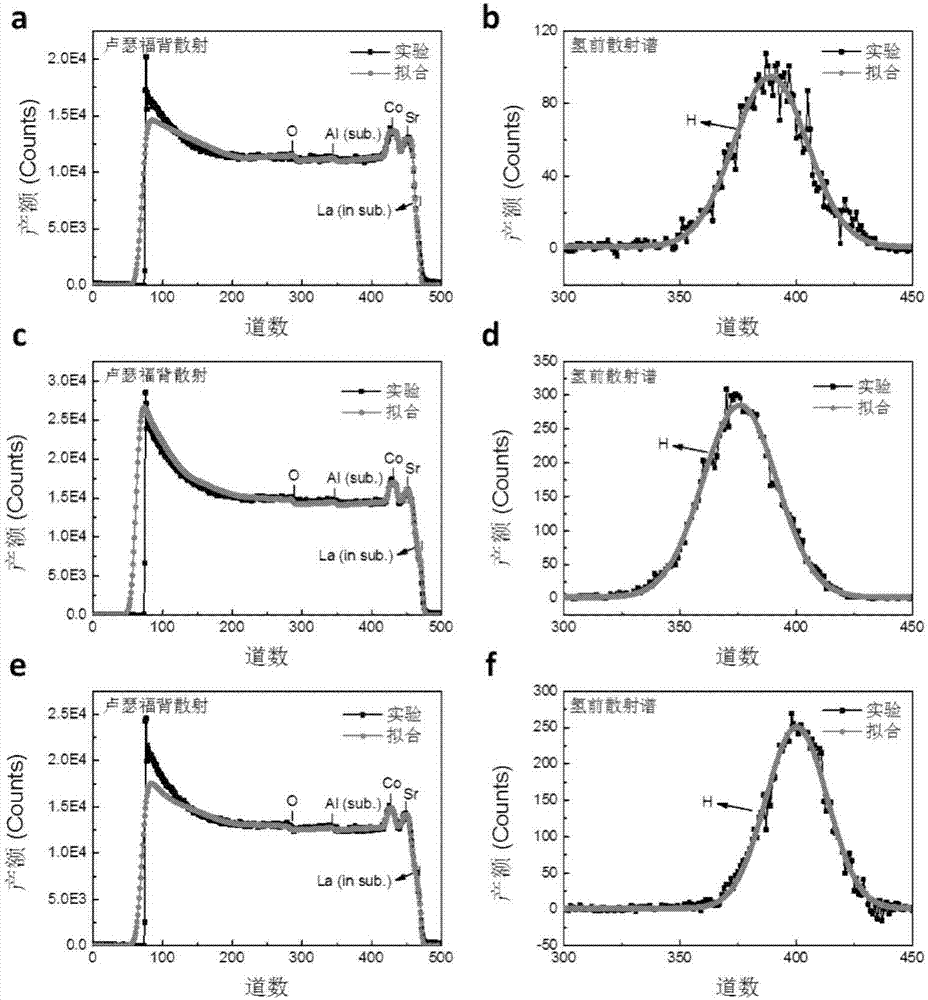
图2为本发明实施例提供的srcoo2.8h0.82(a,b)、srcoo3h1.95(c,d)、srcoo2.5h2.38(e,f)的卢瑟福背散射(rbs)与氢前向散射谱(hfs)的测试曲线;
图3为本发明实施例提供的离子液体栅极电压调控方法的装置及原理图;
图4为本发明实施例提供的原位离子液体栅极电压调控方法中xrd的衍射峰的变化,其对应的相分别为srcoo2.5、srcoo3-δ、srcoo2.5h;
图5为本发明实施例提供的srcoo2.5、srcoo3-δ、srcoo2.5h的x射线衍射的结构表征图;
图6为本发明实施例提供的离子液体栅极电压调控前后薄膜结晶质量的表征;
图7为本发明实施例提供的具有不同厚度的三个相的xrd,即分别对应(a)20nm,(b)40nm,(c)60nmand(d)100nm;
图8为本发明实施例提供的srtio3(001)(a)和laalo3(001)(b)具有不同应力基底上srcoo2.5相的离子液体栅极电压调控后的非原位xrd结果;
图9为本发明实施例提供的三个结构相对应的从xrd形式得到的赝立方晶格体积;
图10为本发明实施例提供的三个相srcoo2.5,srcoo3-δ和srcoo2.5h的co的l带边(a)和o的k带边(b)吸收谱;
图11为本发明实施例提供的用二次离子质谱测量的三个相srcoo2.5,srcoo3-δ和srcoo2.5h中h原子和al原子浓度的深度依赖关系;
图12为本发明实施例提供的新相aboxhy制备方法及三相之间的调控方法;
图13为本发明实施例提供的电致变色三个结构相实物图及其光学带隙的变化;
图14为本发明实施例提供的电致变色三个结构相不同透射谱及其智能玻璃原理图;
图15为本发明实施例提供的从透射谱得到的光学吸收谱;
图16为本发明实施例提供的三个结构相srcoo2.5,srcoo3-δ和srcoo2.5h的电输运特性,即电阻率的温度依懒性;
图17为本发明实施例提供的三个结构相的磁学表征;
图18为本发明实施例提供的具有反铁磁绝缘体特性的srcoo2.5、铁磁绝缘体特性的srcoo2.5h和铁磁金属特性srcoo3-δ三个结构相之间的多态磁电耦合效应;
图19为本发明实施例提供的不同温度下不同磁基态相变所对应的磁电耦合;
图20为基于磁电耦合效应和自旋阀结构构建的五态存储模型展示;
图21为本发明实施例提供的原电池的结构示意图;
图22为本发明实施例提供的原电池的工作原理示意图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例对本发明含氢过渡金属氧化物及其制备方法进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本发明实施例,提供一种能够实现三相相变调控的含氢过渡金属氧化物。所述含氢过渡金属氧化物的结构式为aboxhy,其中a为碱土金属和稀土族元素中的一种或多种,b为过渡族金属元素,x的取值范围为1-3,y的取值范围为0-2.5。其中a与b在aboxhy中的比例不一定是严格的1:1,可以因为存在氧化物中普遍存在的空位和填隙原子等而产生偏离。因此,a与b的比例接近1:1的所述含氢过渡金属氧化物均在本发明保护范围之内。优选地,所述x的取值范围为1-3,y的取值范围为10-2.5。所述碱土金属包括be、mg、ca、sr、ba。所述稀土金属元素包括la、ce、pr、nd、pm、sm、eu、gd、tb、dy、ho、er、tm、yb。所述过渡族元素包括co、cr、fe、mn、ni、cu、ti、zn、sc和v中的一种或多种。可以理解,a还可以是碱土金属与稀土金属的合金,b还可以是过渡金属和主族金属的合金。
所述含氢过渡金属氧化物aboxhy在常温下具有稳定的晶体结构,并且可以在常温下利用离子液体栅极电压调控的方法,通过电场的作用在浸入离子液体中的所述含氢过渡金属氧化物来实现加氢和减氢以及加氧和减氧。进而可以实现从第一相到第二相的相变和从所述第二相返回所述第一相的相变;从所述第一相到第三相的相变和从第三相到第一相的转变;以及从所述第二相到第三相的相变和所述第三相返回所述第二相的相变。其中,所述第一相的晶格体积大于所述第二相的晶格体积,所述第二相的晶格体积大于所述第三相的晶格体积。当然,可以理解还可以通过所述离子液体栅极电压调控的方法,实现上述三个相的循环变换。由于在所述三个相中,所述含氢过渡金属氧化物的物理性质不同,可以通过上述三相的转换实现在电学器件上的应用。所述三个相中的材料的分子式是不同的,第一相中材料为所述含氢过渡金属氧化物aboxhy。所述第二相是在所述含氢过渡金属氧化物aboxhy的基础上,通过所述离子液体栅极电压调控方法,从所述含氢过渡金属氧化物aboxhy析出氢或者注入氧实现。所述第三相是在所述含氢过渡金属氧化物aboxhy的基础上,通过所述离子液体栅极电压调控方法,使得所述含氢过渡金属氧化物aboxhy在第二相基础上近一步析出氢或者注入氧来实现。在一个实施例中,所述三相变换是实现从aboxhy到abo2.5以及abo3-δ的三相之间的变化。同时上述相变可以在电场控制下在三个完全不同相之间形成可逆的结构相变。并且这三个相具有完全不同的电学、光学和磁学特性。以下详细说明本发明的含氢过渡金属氧化物及其制备方法,三相相变,以及应用。
请参见图1,本发明实施例进一步提供一种含氢过渡金属氧化物的制备方法,包括以下步骤:
s100,提供一种结构式为aboz的过渡金属氧化物,其中,其中z大于等于2且小于等于3;
s200,将所述过渡金属氧化物浸入离子液体中;
s300,对所述过渡金属氧化物施加电场,使离子液体中的氢离子插入所述过渡金属氧化物中。
步骤s100中,a为碱土金属和过渡族元素中的一种或多种,b为过渡族金属元素co、cr、fe、mn、ni、cu、ti、zn、sc、v中的一种或多种。所述碱土金属包括be、mg、ca、sr、ba。所述稀土金属元素包括la、ce、pr、nd、pm、sm、eu、gd、tb、dy、ho、er、tm、yb中的一种或多种。所述结构式为aboz的过渡金属氧化物的结构不限,可以是薄膜、粉末、体材、纳米颗粒或者与其他材料的复合材料。在一个实施例中,所述结构式为aboz的过渡金属氧化物为薄膜。可以理解所述过渡金属氧化物为薄膜的制备方法不限,可以采用各种方法制备。
在一个实施例中,所述步骤s100包括以下步骤:
s110,提供基底;
s120,在所述基底表面沉积形成结构式为aboz的过渡金属氧化物薄膜;
s130,在所述过渡金属氧化物薄膜的表面形成第一电极。
所述基底不限,可以为陶瓷基底、硅基底、玻璃基底、金属基底或者聚合物中的一种,只要可以用于成膜的基底都可以应用于步骤s110中。形成所述结构式为aboz的过渡金属氧化物的薄膜的方法不限,可以是各种成膜方法,如离子溅射法、化学气相沉积法、磁控溅射法、凝胶法、激光脉冲沉积等。在一个实施例中,所述步骤s120通过脉冲激光沉积的方法在所述基底上外延生长获得所述过渡金属氧化物薄膜。生长的过渡金属氧化物薄膜厚度不限,优选地所述过渡金属氧化物薄膜的厚度为5纳米至200纳米。所述步骤s130中,所述第一电极与所述过渡金属氧化物薄膜接触形成底电极。可以理解,所述第一电极的位置可以是所述过渡金属氧化物薄膜靠近所述基底的表面,也可以位于所述过渡金属氧化薄膜远离所述基底的表面。所述第一电极可以为金属或者各种导电薄膜以及所述过渡金属氧化物薄膜本身,在一个实施例中所述第一电极为ito薄膜。所述离子液体可以是各种类型的离子液体,在一个实施例中所述离子液体为deme-tfsi。
所述步骤s200中,可以在所述过渡金属氧化物的表面形成一层离子液体层。所述离子液体可以是各种类型的离子液体,只要能够通过水解或者其它途径提供所需氢和氧离子并将所述过渡金属氧化物覆盖即可。当将所述过渡金属氧化物和所述离子液体处于电场中时,可以通过电场的方向来控制离子液体中的氢离子和氧离子插入所述过渡金属氧化物中或者反之析出。所述离子液体中的水的量不限制,实验证实,只要所述离子液体中具有少量的水(>100ppm)就可以实现上述氢离子和氧离子的移动。
可以理解,所述步骤s300中,给所述过渡金属氧化物施加电场的方法可以有多种,在一个实施例中,所述步骤s300包括以下步骤:
s310,提供第二电极以及电源;
s320,将所述第二电极与所述第一电极间隔设置并分别与所述电源电连接;
s330,将所述第二电极浸入所述离子液体中,并通过所述电源施加从所述第二电极向所述第一电极方向的电场。
所述步骤s310中,所述第二电极的形状不限,可以是平行板电极、棒状电极、金属网电极。在一个实施例中,所述第二电极为弹簧状金属丝构成的电极。所述电源可以为各种直流、交流电源等。所述电源的电压可调,可以用来控制反应的时间。在一个实施例中,所述电源为直流电源。
所述步骤s320中,所述第二电极与所述第一电极间隔相对设置,从而可以在所述第二电极与所述第一电极之间形成定向的电场。所述第二电极、所述第一电极与所述直流电源的连接方式不限,可以通过开关控制对所述第二电极以及所述第二电极施加电压。
所述步骤s330中,所述第二电极浸入所述离子液体中,当给所述第一电极以及所述第二电极通电时,可以使得所述第一电极接直流电源的负极,所述第二电极接直流电源的正极。从而可以在所述第一电极与所述第二电极之间产生由所述第二电极向所述第一电极方向的电场。由于所述第一电极与所述第二电极之间具有离子液体,在电场的作用下离子液体中的带正电的氢离子将会向着所述第一电极的方向移动,从而聚集在所述过渡金属氧化物薄膜的表面,进一步插入所述过渡金属氧化物中,从而获得含氢的过渡金属氧化物。而带负电的氧离子将从样品中析出,注入到离子液体中。可以理解,而当翻转电场时候,上述离子变化过程将实现相应的反转。因此,在电场方向变化下,上述过程是可逆过程。
通过所述的离子液体栅极电压调控方法,可以得到具有不同氢和氧含量的钴酸锶的薄膜srcooxhy。在一个实施例中,所述含氢过渡金属氧化物aboxhy为srcoo2.8h0.82、srcoo2.5h、srcoo3h1.95、srcoo2.5h2.38。
请参见图2,为了确定通过上述方法获得的srcooxhy薄膜中h和o的含量,本实施例通过氢前向散射谱(hydrogenforwardscattering)结合卢瑟福背散射(rutherfordbackscattering)的方法对三种srcooxhy薄膜中的h和o含量进行了定量的测量。根据测量结果,得到多个不同薄膜中co和h原子的比例分别为1:0.82(图2a和2b),1:1.95(图2c和2d)和1:2.38(图2e和2f)。并且所述三种srcooxhy的元素化学计量比为srcoo2.8h0.82、srcoo3h1.95和srcoo2.5h2.38。上述srcoo2.8h0.82、srcoo2.5h、srcoo3h1.95、srcoo2.5h2.38均具有在可逆电场控制下实现三个完全不同相之间的拓扑相变,并且这三个结构相具有完全不同的电学、光学和磁学特性。所述含氢过渡金属氧化物aboxhy为srcoo2.8h0.82、srcoo2.5h、srcoo3h1.95、srcoo2.5h2.38。
下面以srcoo2.5h为例,介绍srcoo2.5、srcoo3-δ、srcoo2.5h三相之间的相变。其中srcoo2.5h对应的是第一相,srcoo2.5对应的是第二相,srcoo3-δ对应的是第三相。
请参见图3,栅极电压调控srcoo2.5h相变的装置。通过图3中的装置,采用了离子液体栅极电压调控的方法,实现了在室温下电场控制新相srcoo2.5h的制备以及三个结构相之间的可逆、非挥发性转换。图3中,在所述srcoo2.5h薄膜的边缘涂上银导电胶作为电极,并且所述srcoo2.5h薄膜表面被离子液体覆盖。与所述银导电胶间隔设置的螺旋形的pt电极作为另外一个电极。本实施例中,采用了deme-tfsi型号的离子液体,其可以通过水解其中的水分子得到相变所需要的氢离子和氧离子。但该效应可以推广到其它离子液体、离子盐、聚合物以及极性材料等,只要可以从中获得所需的氢离子和氧离子并使之能够在电场驱动下注入到材料中或者从材料中析出即可。
请参见图4,此图展示的是栅极电压调控法控制三相转变的原位xrd。如图所示,在离子液体中,当对srcoo2.5薄膜采用正的栅极电压后(电压的增加速率是2mv/s),在45.7度(004)的衍射峰逐渐减弱并最终消失。同时,对应新相的44度衍射峰开始出现,从而获得了新的结构物相srcoo2.5h。一旦逐渐变成负的栅极电压,新相srcoo2.5h又很快变回srcoo2.5,并且当一直加负的栅极电压时,srcoo2.5h会转换为具有钙钛矿结构的srcoo3-δ相。此外,这一原位电场调控结构相变可以进行可逆调制。当变为正的栅极电压时,srcoo3-δ相又会快速的变回srcoo2.5相以及srcoo2.5h。因此,通过电场控制的方式,实现了具有钙铁石结构的srcoo2.5、钙钛矿结构的srcoo3-δ和srcoo2.5h相之间的可逆结构相变。更重要的是,这些调控的新相具有非挥发的特性;即撤掉电场后,其结构相及相应的物理特性仍然会保持。
请参见图5,三个结构相srcoo2.5、srcoo3-δ和srcoo2.5h的x射线衍射图片。相比较于具有钙钛矿结构的srcoo3-δ,可以看到具有钙铁石结构的srcoo2.5相展现了一系列源于氧八面体和氧四面体在面外方向的交替排列的超结构峰。基于相应的布拉格衍射角度,srcoo2.5和srcoo3-δ结构的赝立方c轴晶格常数分别是是0.397nm和0.381nm。新相srcoo2.5h也具有一系列的超结构衍射峰,表明srcoo2.5h与srcoo2.5结构具有一样的长程周期性的晶格结构。新相srcoo2.5h的c轴晶格常数是0.411nm,其要比相应的srcoo2.5和srcoo3-δ大出3.7%和8.0%。另外,参见图6,这三个结构相srcoo2.5,srcoo3-δ和srcoo2.5h几乎相同的摇摆曲线半高宽(fwhm)及其和基底相同的面内晶格常数(倒易空间的面内q值是一致的)表明原位生长和栅极电压调控过后薄膜依旧保持高的结晶质量。另外,请参见图7和图8,提供了生长在lsat(001)上具有不同厚度的薄膜(从20nm到100nm)及生长在sto(001)和lao(001)基底上具有不同应力的薄膜,都得到了相似的结果,这充分表明了电场控制的srcoo2.5、srcoo3-δ和srcoo2.5h三相可逆相变的有效性和本质性。即该效应与应力无关,与材料厚度、尺寸无关,能够推广到各种结构形式的材料体系。
请参见图9,从xrd测量得到的三种结构以及其与已有的srcoo3和srcoo2.5体材料的晶格体积对照。从图9可以看出,所述第一相的晶格体积大于所述第二相的晶格体积,所述第二相的晶格体积大于所述第三相的晶格体积。
请参见图10,为了深入理解形成新相srcoo2.5h的电子结构,对三个结构相srcoo2.5,srcoo3-δ和srcoo2.5h中co的l吸收边和o的k吸收边进行了x射线吸收谱测量。co的l2,3吸收边探测的为其电子从2p轨道到3d轨道的的跃迁,可以作为判断相应化合物价态的依据。如图10a所示,从新相srcoo2.5h到srcoo2.5再到srcoo3-δ相,co的l吸收边的峰位逐渐向高能端移动,表明其中co的价态是依次增加的。特别是,新相srcoo2.5h的吸收谱特性和coo具有几乎相同的谱形和峰位,这表明新相srcoo2.5h中co的价态是+2价的。同时,srcoo2.5相中的co的x射线吸收谱也和以前的研究符合的很好,也就是说srcoo2.5相中的co是+3价的。相比于srcoo2.5相,srcoo3-δ相中的co的l3吸收边的峰位要高约0.8ev,表明srcoo3-δ相中具有较少的氧空位(δ<0.1)。此外,通过测量o的k吸收谱对三个结构相的电子态进行了进一步的研究(图10b),其中,o的k吸收测量的是从o1s占据轨道到未占据的o2p轨道之间的跃迁。相比较于srcoo3-δ中o的k吸收边,在srcoo2.5相中,在527.5ev峰位的明显减弱和528.5ev峰位的显著增强表明其从完全的氧八面体配位到部分氧八面体和部分氧四面体配位的转变。然而,在新相中,位于528ev吸收峰的完全消失表明o和co之间的杂化已经很大程度的减弱。
请参见图11,为了证实srcoo2.5晶格中氢离子的插入,采用二次离子质谱的方法测量了三个结构相中h元素和al元素(来源于lsat基底)的深度依赖曲线。相比lsat基底和其它两相,新相中显著的h信号清晰的表明相当数量的h原子已经插入到了srcoo2.5的晶格中,并且其在新相中是均匀分布的。又根据前面吸收谱的测试可以确定co离子的价态+2价的实验证据,可以确定新相的化学式为srcoo2.5h。另外,在ok前置吸收边中(图11b),位于532.5ev很强的吸收峰可归因于o-h键,这也为新相中h+离子的存在提供了很强的证据。
请参见图12,总结了离子液体栅极电压调控的过程及其对三个物相的可逆调控。该结构中,srcoo3具有钙钛矿结构,其中co离子由氧离子所包围形成氧八面体结构。srcoo2.5具有钙铁石结构。相比于srcoo3由于每两个co离子中失去一个氧离子,所以材料形成八面体和四面体的交叠排列。而在srcoo2.5h中,氢离子和氧四面体中的氧离子相连接,形成oh键。这三个结构之间可以通过电场驱动下的氧离子和氢离子的插入和析出实现可逆的结构相变。
请参见图13,提供了电致变色三相实物图及其能隙的变化。请参见图图13a,分别展示了生长在lsat(001)基底上,具有50nm厚度的srcoo2.5,srcoo3-δ和srcoo2.5h三个不同相之间透光度的实物比较。其中srcoo2.5h对应的是第一相,srcoo2.5对应的是第二相,srcoo3-δ对应的是第三相。从图13a可以看出上述三个结构相的透光度的大小。可以发现,srcoo2.5h和lsat(001)基底表现为无色,srcoo2.5表现为褐色,而srcoo3-δ相则表现为黑色。结合电场控制的结构相变,可以发现这一方法可以成为实现电致色变效应的一种非常有效的手段。为了更直观地区分三个结构相之间不同的光学吸收特性,图13b展示的是三个结构相的直接带隙。通过(αω)2-ω公式的拟合,可以发现对比具有金属特性的srcoo3-δ和半导体特性的srcoo2.5(直接带隙2.12ev)。具有co2+的新相srcoo2.5h的直接带隙达到2.84ev,其中的插图也清晰得展现了该结构相变过程中相对应的带隙变化。
请参见图14a中相应的光学透射谱,三相相变中具有双波段的电致变色效应也清晰的得到展现。在可见光区域,srcoo2.5h相(第一相)的透射率要比其它两相高30%以上,而在红外区域(波长达到8000nm),srcoo2.5h相(第一相)和srcoo2.5相(第二相)的透射谱比srcoo3-δ相(第三相)高出60%。另外,图14b展示了由对红外和可见光波段调控所产生的透过度和热效应的不同,即智能玻璃的原理。结合电场控制的的可逆结构相变,当前的srcoo2.5h为电致色变提供了巨大的应用前景,也就是可以通过调控门电压的方式在红外波段和可见光波段有选择性和独立性的进行光透过性的电场调控。具体地,比如在第一相(srcoo2.5h相)时,由于红外和可见光部分的的透过率都比较高,就可以实现同时较多的红外线和可见光进入室内,从而使得室内温度较高、亮度较大。而在第二相(srcoo2.5相)时,由于可见光部分的明显吸收,可以实现室内亮度低但温度较高。而在第三相(srcoo3-δ相)时,由于可见光和红外波段的同时吸收,可以实现室内亮度低并且温度较低。因此,本发明的材料实现了三相相变,拓宽了智能玻璃的应用范围。
请参见图15,为本发明实施例的材料的三相透射谱中得到的光学吸收谱的吸收系数比较。从中可以看出在低于光子能量4.0ev的能量范围内,对所有的三个结构相都存在着两个主要的吸收峰,即低能端的d-d带内跃迁(α,σ和δ)和高能端的p-d带间跃迁(β,ε和γ)。srcoo3-δ在全谱段都表现出较强的光吸收,和其金属特性一致。另外,srcoo2.5和srcoo2.5h都表现为绝缘体特性,其在直接带隙附近形成很强的吸收(β和ε)。另外,srcoo2.5相的光吸收在大于直接带隙的能量区间内甚至强于srcoo3-δ相,这可以归结为srcoo2.5相中较大的p-d跃迁。但是,对于srcoo2.5h相来说,随着直接带隙的增大,吸收被强烈的压制。
请参见图16,透射谱的调制可以理解为源于三个不同相之间的能带结构的不同,其在电输运上也会有所体现。图16展示了三个结构相的电阻率随温度的依赖关系,从中可以看到,srcoo3-δ是很好的金属,其电阻率大约为200uω·cm,而对于srcoo2.5和srcoo2.5h两相,均表现为半导体行为,其室温电阻率分别为8ω·cm和450ω·cm。插图展示的是电场调控下三个结构相之间不同电阻态之间的可逆变化,即中间阻态→高阻态→中间阻态→低阻态→中间阻态。因此,本发明所实现的多阻态之间的电场可控相变构建了基于阻变存储的模型器件单元。
请参见图17,展示了与结构相变密切关联的三态磁电耦合现象,即可以通过电场对于材料的磁性进行调控,从而实现多态磁存储。通过宏观磁性测量,得到srcoo3-δ相的饱和磁矩是2.4μb/co,其居里温度是240k,而srcoo2.5只表现为材料本征的反铁磁行为。另外,图17中srcoo2.5h相也表现出明显的磁滞回线,其饱和磁矩是0.6μb/co,居里温度是125k。
请参见图18,该图示意了由电场控制氧离子和氢离子注入/析出所导致的三个电学和磁学状态之间的调控,其为由电场控制磁性的下一代电子器件提供了新奇和具有潜在应用价值的三态磁电耦合机制。
请参见图19,该图展现了通过电场控制相变或者co的价态,从而实现在不同温度下磁性间的转变。如当温度低于125k时,可以实现从铁磁-反铁磁-铁磁的转变;而在125k-250k之间,则可以实现从铁磁-反铁磁-顺磁的转变;在250k-537k,则可以实现顺磁-反铁磁-顺磁的转变。实际应用中,可以通过电场控制离子移动或者电场控制相变的方法实现不同温度下不同磁基态之间的切换,从而大大丰富了电控磁的范围和内容。
请参见图20,基于三相磁基态的调控,根据其磁电耦合及自旋电子学效应构建了五态存储的模型。通过利用具有不同自旋基态的srcooxhy三相作为自旋固定层,外延磁性金属作为自旋自由层构建自旋阀结构。当调控栅极电压磁基态时,可以实现高组态、低阻态-ⅰ和低阻态-ⅱ,其中低阻态中又会区分出高、低阻态,所以最终可以实现五态存储。
请参见图21,本发明实施例进一步提供一种原电池100,包括间隔设置的阴极电极110和阳极电极130、以及设置与所述阴极电极110和阳极电极130之间的电解质120。所述阴极电极110和所述阳极电极130为本发明实施例提供的含氢过渡金属氧化物的结构式为aboxhy。所述电解质120不限制,只要是现有的具有足够的氧离子和(或)氢离子电导的电解质都可以使用。
请参见图22,所述原电池100的工作原理如下图21所示,在可逆相变的基础上,所述原电池100为基于氢化产物(第一相)和氧化产物(第三相)作为电极材料的原电池。在放电过程中,所述阳极电极130和所述阴极电极110分别放出h+离子和o2-离子,h+离子和o2-离子结合形成h2o,同时伴随有电流产生。在充电过程中,在电场作用下,h2o会发生水解,所述阳极电极130和所述阴极电极110会可逆地发生氢化和氧化,重新生成原来的产物。所以在相变可逆的基础上,所述原电池可以实现可逆充放电。
以上所述实施例仅表达了本发明的几种实施方式,随其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!
- 高氯酸铵内嵌的碳/金属氧化物复合催化剂及其制备方法与制造工艺
- 一种复合氧化物负载杂多酸催化合成尿囊素的方法与制造工艺
- 一种无定形铁锌复合氧化物光芬顿催化剂的制备方法与制造工艺
- 具有杂原子的过渡金属化合物、包含该化合物的催化剂组合物和使用该催化剂组合物的聚合物的制备方法与制造工艺
- 配体化合物、过渡金属化合物和包含该化合物的催化剂组合物的制造方法与工艺
- 一种碳/金属氧化物纳米纤维复合催化剂的制备方法与制造工艺
- 改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与制造工艺
- 一种用于莫来石陶瓷晶须制备的氧化物催化剂的制造方法与工艺
- 一种中空金属蜂窝式氧化催化器的制造方法
- 一种处理汽车尾气的三元金属氧化物催化剂制备方法