具有低接触电阻的有机场效晶体管的制作方法
本发明涉及一种用于具有低接触电阻的场效晶体管的结构,和一种制备该结构的方法。
有机场效晶体管(fet)的效能已在过去十年内由于具有较高电荷载流子迁移率和较高纯度的新材料的开发而极大地改进。fet包括通过由有机材料(us8569746b2)制备的半导体膜分开的一对源极-漏极电极。该膜由栅极电极通过介入栅极介电层控制。适当栅极极性和电压的施加导致载流子在半导体-介电质界面处的累积以得到传导通道。
可取决于栅极电极和源极-漏极电极的相对放置而区分两个重要类别的器件构造:所谓的顶栅极底接触fet和底栅极顶接触fet。通过其注入或收集电荷的半导体与源极-漏极电极之间的接面还称作“接触件”。原则上,两个其他构造(顶栅极顶接触fet和底栅极底接触fet)为可能的,但这些构造通常在接触件与传导通道之间的接面处遭受更严重的电流集聚效应且因此在技术方面具有受限应用。
通过fet的总电阻由通道的电阻(rch)和接触件的电阻(rc)的总和得到:
方程式1.rtol=rch+rc
rch由在半导体-介电质界面处累积的二维载流子密度测定。这取决于栅场且以一级近似由下式得到:
方程式2.
其中vds为源极-漏极电压,vgs为栅极电压,vth为栅极阈值,ids为源极-漏极电流,μfet为场效晶体管迁移率,c为栅极介电质电容,且l和w分别为通道长度和宽度。
另一方面,接触电阻rc为电荷载流子在其由源极电极移动至通道且由通道移动至漏极电极时所遇到的有效电阻。该rc为由使必需源极-漏极电流通过电极与通道之间的半导体区域所需的体电压产生的空间电荷电阻rsclc与接触件的有效电极接触电阻re的总和:
方程式3.rc=rsclc+re
re由有效电极接触电阻率re与有效接触面积ae的乘积得到。文献中通常不区分rc与re。然而,区分rc与re在目前为重要的。re主要取决于电极-半导体接触和器件几何形状的细节。rsclc主要取决于大部分膜中的载流子的空间电荷迁移率、由电极至通道的距离和存在的任何电流集聚效应。
本发明目的在于减小re和因此减小re。预期re取决于所使用的电极(例如金、银、铜等)和是否存在覆盖层,以及与电极的费米能级有关的半导体传输级的能量(例如空穴的最高分子占据轨道(homo)能带边缘和电子的最低分子占据轨道(lumo)能带边缘)。
据说在re远小于rch,优选地小于rch的十分之一,更优选地小于rch的三十分之一时实现欧姆接触的状态。
对于具有约0.5cm2v-1s-1的场效迁移率的有机半导体材料,100μm的器件通道长度的rch在“接通”状态中为300kωcm。这意指必须保持源极和漏极接触件的总rc小于300kωcm的十分之一(即小于30kωcm),从而使接触件不严重限制晶体管的效能,包括速度、带宽和接通特性。在现有技术中这要求很高且不可容易地实现。
已发现具有金源极-漏极电极的顶栅极底接触fet的当前可得rc对包括如下的宽范围的有机半导体为105-106ωcm:并五苯(pn)和6,13-双(三异丙基甲硅烷基乙炔基)并五苯(tips-pn),如在d.boudinet等,organicelectronics,11,227(2010)中所述;和聚[2,5-双(3-十四烷基噻吩-2-基)噻吩并[3,2-b]噻吩](pbttt),如在y.y.noh等,semiconductorscienceandtechnology,26,034003(2011)中所述。所有这些有机半导体具有相对低的离子化电位(ip):pbttt,4.8ev;pn和tips-pn,4.9ev。因此,rc甚至对于具有相对长通道长度的器件且对于具有相对低ip的半导体(可预期其更容易地得到欧姆行为)并非足够低。这随着场效迁移率变得更大和/或通道长度变得更短而进一步降低:容许rc成比例地变小。
广泛已知在有机fet技术中用作空穴-载流子注入电极的典型金属(例如金、银和铜)不具有足够大的功函数来实现与具有大于约5.0ev的ip的有机半导体的欧姆接触。这不幸地涵盖作为p通道材料的目前和未来技术关注的大多数材料。
减小re和减小因此re的关键为降低至半导体中的有效电荷注入能垒。对p通道fet,已尝试数个方法来实现此。
一种方法为通过改性源极-漏极电极的表面以增加其功函数而增加源极-漏极电极的功函数。用于改性金属电极表面以增加其功函数的一种方法使用所谓的自组装单分子层(sam)。campbell等在physicalreviewb,54(2008),第14321页中证实硫醇sam可在组装于银表面时通过偶极机构导致大功函数变化。
其应用于fet的一个实例由asadi等在journalofmaterialschemistry,17(2007),第1947页中给出,其中报道1h,1h,2h,2h-全氟癸硫醇和十六烷硫醇sam分别将金的功函数增加至5.6ev和将金的功函数减小至4.0ev。然而,fet器件对作为半导体的发橙光聚对亚苯基亚乙烯基显示出rc由1mωcm至分别4和7mωcm的增加,且对作为半导体的立体规则性聚(3-己基噻吩)显示出由80kωcm至100kωcm的增加。
另一实例由noh等在semiconductorsciencetechnology,26(2011),第034003页中给出,其中报道1h,1h,2h,2h-全氟癸硫醇sam将au的功函数由约4.4ev增加至5.0ev。发现rc对作为半导体的pbttt由150
在某些情况下,sam似乎促进有利于载流子注入的有机半导体的边界层的更理想组织和/或定向。kawasaki等在japanesejournalofappliedphysics,47(2008),第6247页中描述使用疏水性硫醇封端的sam来改进蒸发pn半导体的定向和形态。
因此,尽管显然某些sam能够增加金属表面的功函数,但其可能由于所引入的额外隧障势垒而通常不显著减小rc。此外,氟化sam形成半导体覆盖层的润湿和溶液沉积的新挑战。最终,硫醇sam尤其在金属表面上具有有限的热和化学稳定性。
用于改性金属电极表面以增加其功函数的第二种方法使用具有高功函数的p掺杂导电聚合物。一个实例由hong等在organicelectronics,9(2008),第864页中给出,其中厚的p掺杂导电聚合物空穴注入层(聚(3,4,-亚乙基二氧噻吩):聚(苯乙烯磺酸)(pedt:pssh))由旋转浇注在金源极-漏极电极数组上方。这利用疏水性通道区域上方的溶液的自发除湿来图案化导电聚合物膜。其发现rc对作为半导体的pn由3mωcm减小至140kωcm。
该方法非常依赖于基材的电极和通道区域上方的厚导电聚合物膜的差动润湿-除湿。在具有小通道长度的器件上实施是非常具有挑战性的,除非引入数个额外光刻步骤。此外,其可仅为具有小于导电聚合物层的功函数的ip的半导体仅提供良好空穴接触,这限制该方法的应用,即使可发现良好光刻方法用于图案化对准至源极-漏极电极数组的导电聚合物膜。
用于改性金属电极表面以增加其功函数的第三种方法在其表面上方形成合适氧化物层。一个实例由kim等在journalofappliedphysics,84(1998),第6859页中给出,其报道ito的功函数由4.5ev增加至4.75evo2等离子体。
用于改性金属电极表面以增加其功函数的第四种方法使用具有高功函数的半导体无机氧化物。一个实例由kumaki等在appliedphysicsletters,92(2008),第013301页中给出,其中moox层和随后的金覆盖层在sio2/si基材上的光致抗蚀剂图案化井中蒸发,然后沉积pn有机半导体以得到底栅极底通道fet。报道rc在使用moox层时由13mωcm减小至240kωcm。
另一实例由gui等在organicelectronics,15(2014),第3349页中给出,其中tio2或经mn掺杂tio2层通过金属掩模和随后的金源极-漏极数组热蒸发至pn有机半导体层(通道)上。报道fet的rc在使用tio2和经mn掺杂tio2时由2mωcm分别减小至40kωcm和30kωcm。
该方法需要进行顺序蒸发以通过阴影或光刻/光致抗蚀剂掩模界定源极-漏极电极。若电极将通过电镀或印刷技术来沉积,则该方法将需要额外光刻步骤以用于对准。
第二种方法引入中间半导体层以减小用于注入有机半导体中的整体势垒。还称作梯度注入方法的方法由ho等在nature,404(2000),第481页中描述。至有机fet的应用由park等在appliedphysicsletters,88(2006),第113503页中给出,其中星爆型胺4,4’,4”-三(n-3-甲基苯基-n-苯基氨基)-三苯胺(m-mtdata)的薄膜插入在顶接触器件中的pn半导体与金电极之间以形成金费米能级与pn的homo边缘之间的中间能级。这将rc由8mωcm减小至1mωcm。
另一实例由chen等在appliedphysicsletters,90(2007),第073504页中给出,其中铜酞菁(cupc)层插入在顶接触器件中的pn与经蒸发金电极之间以形成金费米能级与pn的homo之间的中间能级。这将rc由470kωcm减小至155kωcm。
因此,显然尽管可使用梯度注入方法显著改进rc,但最终rc甚至对具有相对低ip的有机半导体仍可为相对大的。该残余rc可归因于注入能垒的不完整消除(由于步骤的不完美梯度)。
第三种方法在有机半导体与金属电极的接面处引入有机半导体的有意p掺杂中间层,因此p掺杂中间层提供至有机半导体的欧姆接触。实现此的一种方法通过使用与有机半导体共同蒸发或顺序地蒸发的高电子亲和力材料(例如moox及其他氧化剂)而在电极区域处掺杂有机半导体。
该方法的一个实例由li等在superlatticesandmicrostructures,50(2011),第191页中给出,其中pn-moox混合的中间层通过金属掩模和随后的金电极蒸发至pn有机半导体层上。moox在经混合中间层中p掺杂pn。发现ofet的rc在中间层经优化时由60kωcm减小至7kωcm。
另一实例由lin等在ecssolidstateletters,3(2014),第81-83页中给出,其中2,3,5,6-四氟-7,7,8,8-四氰代二甲基苯醌(f4-tcnq)的薄层通过蒸发通过顶接触fet中的阴影掩模插入pn与金之间。f4-tcnq掺杂pn的接触区域。发现rc由44kωcm减小至30kωcm。
第三个实例由vanoni等在appliedphysicsletters,90,193119(2007)中给出,其中f4-tcnq与pn共同蒸发以得到底栅极底接触fet中的经掺杂pn层。发现rc由110kωcm减小至5kωcm。
第四个实例由minari等在appliedphysicsletters,100(2012),第093303页中给出,其中在顶接触fet中的au源极-漏极电极的蒸发之前fecl3通过阴影掩模蒸发以掺杂二辛基苯并噻吩并苯并噻吩膜的接触区域。发现这将rc由200kωcm减小至9kωcm。
因此,显然接触区域的掺杂可显著减小rc。然而,对于在这些现有技术中的方法存在数个关键挑战。通常,这些方法需要通过阴影或光刻掩模的多步骤真空处理。这极大地增加复杂度和成本,且限制可扩缩性(scalability)。其还经不起溶液处理。其次,在具有小分子或无机复合掺杂剂的那些情况下,掺杂剂分子或复合体可在有机半导体内侧随着时间迁移。这最终使接触件处的掺杂轮廓和因此其re降级。因此期望开发可克服这些缺点的掺杂接触区域的替代方法。
用于在电极区域处掺杂有机半导体的第二种方法使用反应性中间层。klauk等在美国专利us6,806,124中描述使用反应性中间层以在源极-漏极电极附近掺杂有机半导体层区域。导电聚合物层(例如p掺杂聚苯胺)与可扩散掺杂剂(例如樟脑磺酸)一起描述。掺杂剂扩散至半导体中且在接触件附近掺杂。该方法中不存在限制掺杂剂至界面的扩散的可能性。当掺杂剂扩散至通道区域时,其导致高背景电导率和不良开关比。
klauk等还描述使用具有结合至金属表面的一个基团和用作用于将空穴转移至有机半导体的氧化剂由此对其进行p掺杂的第二基团的双功能掺杂剂层。引用硫醇作为可能实例。然而,硫醇通常不与掺杂具有大于约5.0ev的ip的有机半导体所需的氧化剂相容。
用于在电极接触件处形成经掺杂半导体层的第三种方法沉积经掺杂半导体自身。一个实例由shih等在美国专利us7208756中且在journalofappliedphysics,96(2004),第454页中给出,该期刊描述在接触件处使用极轻p掺杂的聚合物中间层。由通道材料(例如聚(3-己基噻吩))与低浓度的掺杂剂(例如处于1%以下浓度的fecl3)的混合物沉积该中间层。该轻掺杂的中间层还沉积在通道上方。
因此,该轻掺杂的中间层的电阻率需要经选择使得其电阻比通过通道的接通状态提供的电阻高得多,但还是足够低以减小re。结果为条件要求很高且在大多数情况下无法满足。例如若在相对高迁移率材料(例如具有μfet=0.1cm2v-1s-1的经轻掺杂聚(3-己基噻吩))中期望大于1,000的开关比,则不可能在不牺牲开关比的情况下将rc由180mωcm减小至低于600kωcm。
该方法的另一关键挑战为缺乏提供经掺杂中间层至电极的自对准(避免在通道区域上方沉积导电层)。还不存在在后续溶剂处理、热处理期间、在储存时且在操作期间抑制掺杂剂扩散出中间层和导致的器件效能降低的可能性。
第四种方法由schaur等在organicelectronics,13(2012),第1296页中给出,其描述使用常规电化学以在顶接触底栅极fet中的电极与结晶有机半导体薄膜pn之间的接面处形成浅p掺杂区域。发现rc由390kωcm减小至140kωcm。该方法需要使用电化学且不能够在电极的整个埋入接触件上方产生连续p掺杂层。
依据上文的总结,显然仍需要提供对具有大于5.0ev的ip(例如5.0至5.8ev)的有机半导体可可靠地得到小于30kωcm的re的改进器件结构。进一步需要提供可提供该欧姆接触的层。进一步需要提供用于沉积该层的方法。
本发明公开了通过在电极数组与半导体之间的接面处插入结合阴离子的分子薄层而在源极-漏极电极数组上方提供半导体的超薄层的受控p掺杂的晶体管器件结构。在器件的制备期间首先用氧化剂物质配制该分子薄层。在不因此受理论限制的情况下,氧化剂产生半导体界面的p掺杂。可已经存在或随后沉积半导体层。由界面移除氧化剂副产物,且结合阴离子然后充当至p掺杂界面的抗衡阴离子。该结合阴离子层可取决于器件构造而使用各种方法自对准至电极数组。有限氧化剂连同结合阴离子层一起仅确保在电极上方局部掺杂半导体的超薄层。本发明进一步提供在制备期间采用的含氧化剂层以实现半导体的该受控p掺杂。本发明进一步提供基于溶液处理的自对准方法以产生该层。这产生对具有大于5.0ev的ip的半导体显示出欧姆注入和高性能的场效晶体管,这在先前为不可能的。
在一个实施方案中,晶体管器件结构可包括:
(i)图案化源极-漏极电极数组层;
(ii)结合阴离子的分子薄层,其自对准至源极-漏极电极数组图案,且夹在电极层与半导体层之间;
(iii)半导体层,其具有邻接于结合阴离子层的p掺杂界面;
(iv)栅极介电层;
(v)图案化栅极电极层。
在一个实施方案中,含氧化剂层可包括:
(i)与阴离子且任选地与基材结合基团连接的聚合物或低聚物;
(ii)氧化剂阳离子物质,其相对于标准氢电极具有大于0.5v的标准电极电位。
在一个实施方案中,制备含氧化物层的方法可包括如下:
(i)沉积含有第一阳离子的与阴离子且任选地与基材结合基团连接的聚合物或低聚物的分子薄层;
(ii)交换第一阳离子与氧化剂阳离子。
该方法的关键益处为分离由结合阴离子提供的抗衡阴离子与由氧化剂物质提供的p掺杂剂的作用。因此,氧化剂可适当地经选择以匹配半导体的ip,而结合阴离子层可经选择以粘合至基材且提供p掺杂界面的所需处理、热、储存和操作稳定性。
因此,本发明的第一方面提供在源极-漏极电极数组层与半导体层之间并入结合阴离子的分子薄层的场效晶体管器件结构,其中半导体层在其与该结合阴离子层的界面处p掺杂,且结合阴离子的分子薄层能够结合至基材,在该基材上形成结合阴离子的该分子薄层。
本发明的第二方面提供结合阴离子和氧化剂物质的含氧化剂分子薄层。
本发明的第三方面提供用于制备该层的方法。
本发明的第四方面提供用于制备该场效晶体管结构的方法。
本发明的第五方面提供该场效晶体管结构在电子电路中的用途。
图1中对四个可能器件构造中的每一个显示出器件结构。半导体层在其与对准至源极-漏极电极数组的结合阴离子分子薄层的接触件处选择性地p掺杂。结合阴离子层可取决于器件构造而使用如稍后描述的各种方法自对准至电极数组。这无需单独图案化和对齐步骤。其还确保恰好在需要得到欧姆注入之处形成p掺杂层。
在不因此受理论限制的情况下,该器件结构具有如下关键优点:由于p掺杂半导体中的正电荷由结合阴离子抗衡,因此掺杂轮廓(即随半导体中的位置而变的经掺杂载流子密度)变得通过该结合阴离子层的位置固定。因此,掺杂轮廓理想地限于邻接于结合阴离子层的几纳米厚的超薄层。此外,由于结合阴离子无法扩散,因此掺杂轮廓还无法涂抹且随着时间降低。因此,p掺杂界面对于器件处理、烘烤至中等温度(高达180℃)、储存和操作为稳定的。该器件结构因此对掺杂剂迁移和所导致的掺杂轮廓的降低具有固有免疫。
结合阴离子抗衡最终晶体管结构中的p掺杂聚合物界面的正电荷。然而,结合阴离子层在器件处理的初始阶段中连同氧化剂物质一起沉积,在该器件处理期间结合阴离子由旁观(spectator)阳离子或氧化剂阳离子抗衡。对底接触器件构造,该含氧化剂层在电极数组层之后但在半导体层之前沉积。对顶接触器件构造,该含氧化剂层在半导体层之后但在电极数组层之前沉积。
在不因此受理论限制的情况下,发生由半导体至结合阴离子层中的氧化剂物质的电子转移。这导致半导体界面的氧化(即p掺杂)以及氧化剂物质的对应还原。在该方法期间,自发地消除旁观或氧化剂阳离子,p掺杂聚合物的正电荷由此而变得由结合阴离子抗衡。
因此,最终器件结构中的术语“结合阴离子层”是指由p掺杂聚合物抗衡的阴离子层,然而在半导体的p掺杂之前的中间处理阶段中,其是指由其旁观和/或氧化剂阳离子抗衡的阴离子层。将由其中使用该术语的上下文明了该预期含义。术语“结合阴离子材料”是指沉积以得到结合阴离子层的材料。
结合阴离子的作用为最终平衡界面p掺杂聚合物的正电荷。因此,其不应经历与p掺杂聚合物的化学反应。这需要阴离子应为仅弱亲核的,优选地为非亲核的。亲核性为阴离子参与与为带正电有机半导体的亲电体的结合形成反应的倾向(参见例如:march,advancedorganicchemistry:reactions,mechanismsandstructures,wiley)。合适结合阴离子包括磺酸根、氟烷基磺酸根、羧酸根、氟烷基羧酸根、膦酸根、磷酸根和硫酸根以及其组合,更优选地,磺酸根、氟烷基磺酸根和氟烷基羧酸根。
这些阴离子共价地连接在结合阴离子材料中,优选地连接至合适聚合物或低聚物骨架。聚合物为具有大于5kda且通常大于结合在一起的10个相同或不相似单体单元的相对高分子量的大分子。合适聚合物骨架包括非共轭聚合物骨架,例如聚苯乙烯、聚丙烯酸酯、聚甲基丙烯酸酯、聚(乙烯醇)、聚(烯丙胺)、聚乙烯亚胺;和更优选地共轭聚合物骨架,例如聚二酮基吡咯并[3,4-c]吡咯、聚噻吩、聚(联噻吩-alt-噻吩并噻吩)。进一步优选地,选择类似于用作器件中的半导体的核的共轭核。低聚物为具有5kda或更少且通常为结合在一起的至少两个和至多十个相同或不相似单体单元的相对低分子量的大分子。合适低聚物骨架包括上述的低分子量变体。
阴离子基团以每0.2nm3至4nm3结合阴离子层一个阴离子基团的密度连接至聚合物或低聚物。该连接可通过短烷基链间隔基团。结合阴离子层因此可为均聚物、共聚物/均低聚物或共低聚物。在一个实施方案中,其为聚电解质。聚电解质为具有高离子基团密度(例如每0.2至1nm3一个阴离子基团)的聚合物。在另一实施方案中,结合阴离子层为离聚物。离聚物为具有低离子基团密度(例如每2nm3材料一个或更少阴离子基团)的聚合物。
阴离子基团最初可由旁观阳离子抗衡。旁观阳离子为存在于结合阴离子层中以抗衡阳离子但不提供氧化作用的阳离子。旁观阳离子的实例为锂、钠、钾、铷、铯、铵和季铵。旁观阳离子经选择使得其将合适溶剂加工性提供至结合阴离子层。旁观阳离子随后可由氧化剂阳离子替换,如下文所述。
申请人发现,结合阴离子的超薄层令人惊奇地可充分组装至金属电极表面上以最终将所需电荷抗衡提供至p掺杂半导体界面。结合阴离子层的所需厚度为0.3nm至3nm,更优选地为0.5nm至2nm。将这些层称为“分子薄”的,这是因为其对应于分子尺寸。厚结合阴离子层由于其引入的额外电阻而为较不合适的。
此外,结合阴离子分子薄层可通过自组装容易地形成于基材上,优选地通过自组装形成至预图案化电极表面。以该方式可靠地形成厚层通常为不可能的。
可在初始基材表面的光学介电函数的合适模型化之后对沉积在已知定义的模型基材(例如硅或金)上的情况使用可变角度光谱椭圆偏振法估计所需膜厚度(参见例如:fujiwara,spectroscopicellipsometry:principlesandapplications,wiley(新加坡)中)。还可通过与光电发射的电子的非弹性平均自由程效应有关的熟知的电子衰减效应使用存在于结合阴离子层中的标记原子的核级x射线光电发射光谱法估计该膜厚度(参见例如:briggs和seah,practicalsurfaceanalysisbyaugerandx-rayphotoemissionspectroscopy,wiley(新加坡)中)。
结合阴离子层中的阴离子密度的选择由半导体的界面处的优选p掺杂密度和结合阴离子层的厚度来测定。在不因此受理论限制的情况下,层的厚度(以nm计)与其阴离子密度(以每nm3的阴离子电荷计)的乘积得到总区域阴离子电荷密度和因此结合阴离子层可在半导体界面处支持的最大p掺杂密度(以每nm2的空穴计)。优选p掺杂密度为3×1012至3×1014个空穴/cm2,或更优选地1×1013至1×1014个空穴/cm2。
此外,结合阴离子层可任选地含有基材结合基团。申请人惊奇地发现,某些阴离子基团(例如磺酸根和膦酸根)能够非特定地以通常方式结合至金属电极表面,例如金、银和铜。然而,其不结合至通常形成金属电极的基材的疏水表面,例如由聚(对苯二甲酸乙二醇酯)、聚(萘二甲酸乙二醇酯)、聚酰亚胺、聚碳酸酯和hmds处理的sio2晶片制备的塑料箔片。
在不因此受理论限制的情况下,高度可极化阴离子基团连同其抗衡阳离子一起据信通过伦敦分散机制提供比有机表面强的与金属表面的范德华交互作用。这实现结合阴离子层至电极表面上的选择性组装。因此,可实现该层至下面的图案化电极数组层的自对准。这对于底接触fet器件可为尤其有用的。
金属电极的合适结合阴离子层的实例为聚苯乙烯磺酸盐聚电解质类。这包括聚(苯乙烯磺酸)及其与锂、钠、钾、铷、铯、铵和四甲基铵的盐。
合适结合阴离子层的另一实例为具有由自由阳离子抗衡的总阴离子结合电荷的聚电解质复合体。聚电解质复合体为包括聚阳离子和聚阴离子的材料。若聚阳离子中的阳离子电荷小于补偿聚阴离子中的阴离子电荷所需的数目,则聚电解质复合体为整体阴离子的。合适聚电解质复合体的实例为聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸)(pedt:pssh)。合适结合阴离子层的另一实例为具有相对于结合阳离子电荷过量的结合阴离子电荷的阴离子聚两性电解质。
合适结合阴离子层的其他实例包括共轭聚电解质聚(n,n-双(3-磺酸基丙基)-1,4-二酮基吡咯并[3,4-c]吡咯-3,6-二基-alt-三联噻吩铯)和聚(3-磺酸基丁基噻吩-2,5-二基钾)。
然而,可能进一步有利的是并入可结合至金属电极表面的额外特定结合基团。实例包括异羟肟酸及其盐类、儿茶酚及其盐类、噻吩、吡啶和胺。这些基团可作为结合阴离子材料中的侧基以低浓度(优选地5至20mol%(以重量计))并入。
为了结合至顶接触器件中的有机半导体膜基材,结合阴离子材料可优选地具有可结合至所需有机半导体膜的基材结合基团。在不因此受理论限制的情况下,这些基团可与半导体形成供体-受体复合体。这些基团包括:可结合至富电子有机半导体的吸电子基团,例如二硝基苯基、二氰基苯基、全氟苯基和多氯萘基以及其衍生物;和可结合至缺电子有机半导体的给电子基团,例如二烷氧基苯基、二羟基苯基、二氨基苯基及其衍生物。结合基团的其他实例为得到与有机表面的非特定交互作用的那些结合基团。这些包括全氟烷基和全氟烷氧基链。这些基团可作为结合阴离子材料中的侧基以低浓度(优选地5至20mol%(以重量计))并入。
含氧化剂结合阴离子层含有起半导体的p掺杂作用的一种或多种氧化剂物质。氧化剂物质为可氧化(即p掺杂)期望半导体的任何合适低分子量试剂。因此,这还称作p掺杂剂。
合适氧化剂为还为阳离子的单电子氧化剂。单电子氧化剂引起一个电子的转移和因此使半导体掺杂有每氧化剂分子或离子一个空穴。这可通过离子交换并入结合阴离子材料中。这还可通过以下方式并入结合阴离子层中:通过阳离子交换机制使该层与这些氧化剂阳离子的合适溶液接触。优选地,氧化剂阳离子还为单带电阳离子。在该情况下,p掺杂导致氧化剂形成可有利地在处理期间由结合阴离子层移除的不带电副产物。
可用作单电子氧化剂的单带电阳离子的实例包括(相对于标准氢电极she,标准电极电位e°):三(对硝基苯基)铵(1.84v)、三(2,4-二溴苯基)铵(1.78v)、三(对氰基苯基)铵(1.72v)、硝
合适氧化剂物质还可为可p掺杂半导体的任何氧化剂物质。实例包括过二硫酸盐(2.1v)和过氧化氢(1.8v)。这些氧化剂可通过以下方式并入结合阴离子层中:使该层与这些氧化剂的合适溶液接触。
氧化剂的选择标准为其标准电极电位e°。e°为在其下氧化剂的单位活性与其经还原物质的单位活性相平衡的电极电位。这可通过伏安法而容易地测量(参见例如:faulkner和bard,electrochemicalmethods:fundamentalsandapplications,wiley(新加坡))。更正性e°指示更强氧化剂。在不因此受理论限制的情况下,以下说明可为有用的。所需氧化剂应优选地具有比(ip-4.56v)正约0.3v的e°(相对于she),其中ip为半导体膜的离子化电位。
进一步有利的是选择仅足够强以p掺杂半导体的氧化剂。更强氧化剂在周围环境中趋于不稳定。
半导体的离子化电位为由固体膜中的半导体的homo边缘移除电子所需的最小能量。这可通过紫外线光电发射光谱法(ups)由光电发射的开始与由光谱中的低动能截止得出的真空能级之间的能量差得到。
对底接触器件,合适结合阴离子层通过自组装沉积在源极-漏极电极表面上。这可通过使用旋涂、滴涂、喷墨印刷或其他溶液处理方法使基材与所需结合阴离子材料的合适溶剂中的稀释溶液接触实现。可通过实验得出所需溶液浓度(通常为0.25-10mg/ml)。可需要室温或稍微升高温度下的短接触时间(1-5min)。然后用合适溶剂移除过量材料。合适溶剂为使结合阴离子材料溶解的任何溶剂。中等低温下的短烘烤可用于进一步改进结合阴离子层至电极表面的粘合。本领域熟练技术人员可测定精确条件。
结合阴离子层选择性地结合至源极-漏极电极数组而非电极之间的疏水性空间,即通道区域。以该方式,实现至电极图案的自对准。
氧化剂可并入结合阴离子材料中或可随后引入所沉积结合阴离子层中。为进行后一操作,使合适溶剂中的氧化剂的溶液(通常10至50mm)与结合阴离子层接触短时间周期(通常1至30s)且然后通过在溶剂浴中旋转或浸洗而移除溶液。然后通过合适溶剂移除过量氧化剂。合适溶剂为使氧化剂材料而非结合阴离子层溶解的溶剂。
然后通过合适溶剂沉积半导体覆盖层。这产生与充当抗衡阴离子的结合阴离子的p掺杂界面。可通过用于沉积半导体的溶剂同时移除经还原氧化剂副产物。在某些情况下,可需要温和烘烤步骤以活化p掺杂(通常为100至120℃,1分钟)。
若需要,则可通过对氧化剂物质在超薄膜中的存在敏感的任何方法验证氧化剂物质在结合阴离子层中的存在。这包括x射线光电发射光谱法。在其中氧化剂物质显示出共振增强拉曼散射剖面的情况下,还可使用拉曼光谱法。
对顶接触器件,合适结合阴离子层通过自组装沉积在现在充当基材的半导体表面上。这可通过使基材与合适溶剂中的稀释溶液接触以及如上文所述的处理而实现。可如上文所述并入氧化剂。有利的是选择不允许氧化剂物质渗透超过结合阴离子层的氧化剂物质和溶剂。这意指对于下面的半导体,氧化剂物质应优选地较大,且溶剂应优选地为不良溶剂。可如上文所述验证结合阴离子层的存在。
在该方法中,将半导体底层与充当抗衡阴离子的结合阴离子同时p掺杂。然后可通过阴影掩模通过蒸发沉积源极-漏极电极数组层。然后可通过现在充当掩模的源极-漏极电极数组通过与合适还原剂(例如肼蒸汽或短氧等离子体)的短暂接触而将通道区域中的p掺杂失活。这因此产生自对准至电极数组的p掺杂界面。
因此对顶栅极底接触器件,首先沉积图案化源极-漏极电极数组层,然后自对准沉积并入氧化剂物质的结合阴离子层,然后沉积半导体层、栅极介电层和图案化栅极电极层。
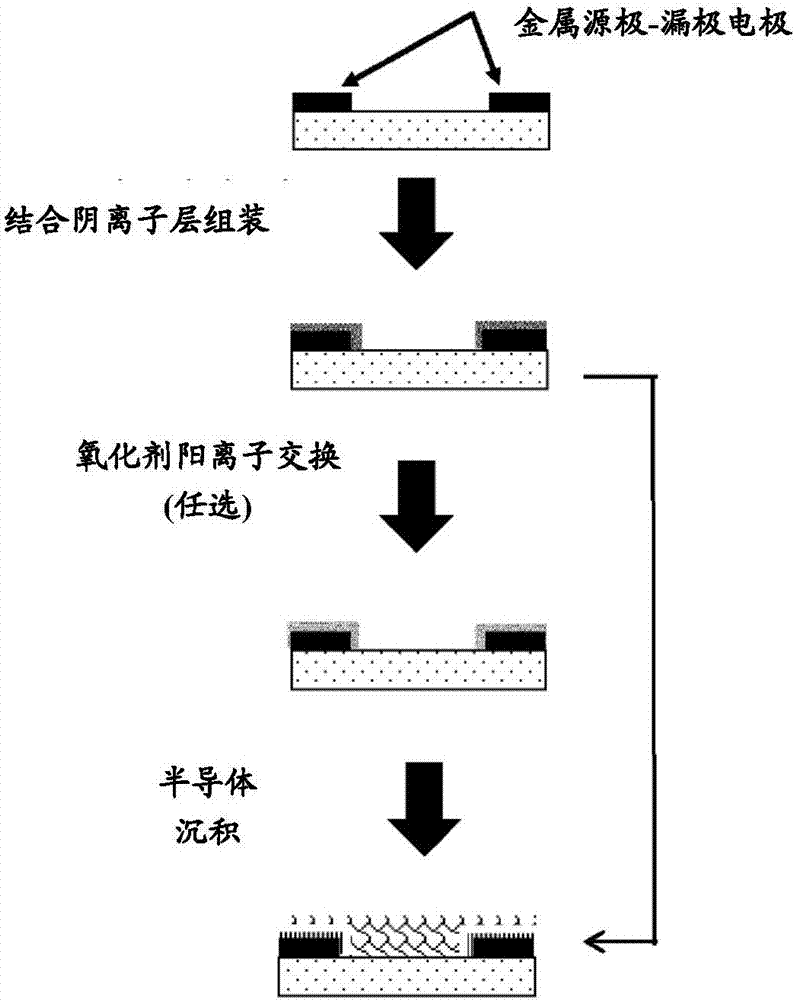
对底栅极底接触器件,首先沉积图案化栅极电极层,然后沉积栅极介电层、图案化源极-漏极电极数组层,然后自对准沉积并入氧化剂物质的结合阴离子层,然后沉积半导体层。图2中示意了一个实施方案。
对底栅极顶接触器件,首先沉积图案化栅极电极层,然后沉积栅极介电层、半导体层、并入氧化剂物质的结合阴离子层,然后沉积图案化源极-漏极电极层,和然后是通道区域中的p掺杂的任选失活。
对顶栅极顶接触器件,首先沉积半导体层,然后沉积并入氧化剂物质的结合阴离子层,然后沉积图案化源极-漏极电极数组层,和然后是通道区域中的p掺杂的任选失活,然后沉积栅极介电层和图案化栅极电极层。
为估计rc,申请人开发了一种自洽传输线方法(tlm),该方法可由随源极-漏极电流和通道载流子密度而变的所测量晶体管电流-电压特性可靠地得到该量。简言之,场效晶体管的转移曲线对栅极泄漏进行收集和校正。源极-漏极电流(ids)然后对不同源极-漏极电压(vds)在选择栅极电压(vgs)下得到以给出ids-vds-vgs表面,且标准化至通道宽度。该表面与多项式函数拟合以在不同ids和vgs下内推vds。然后通过rtot=-vds/ids获得rtot。对不同通道长度重复该程序以得到rtot与l关系曲线图,可由该曲线图作为截距而获得rc。将rc估计为vgs的函数。然后可通过由空间电荷限制的电流的模型化减去rc贡献而获得re。对欧姆接触,器件显示出re对ids和vgs的极小依赖性。然后可通过与由电流的传输线模型自洽地获得的有效电极区域的标准化而由rc获得re。
图9中显示出来自使用中间结合阴离子层psstpa.的改进效能fet器件的接触电阻提取结果的实例。
对模型高迁移率空穴类型聚合物有机半导体(聚(n,n-双(2-辛基十二烷基)-1,4-二酮基吡咯并[3,4-c]吡咯-3,6-二基-alt-三联噻吩)(dppt-t2))测量的金电极的典型re为约20ωcm2,其中ip为5.3ev。这产生类似于或大于rsclc的相当大的re。因此,具有短于50μm的通道长度的典型场效晶体管受接触件限制。使用此处所公开的本发明,申请人已实现低至2ωcm2的re,re为足够良好的以达到用于至半导体膜中的电荷载流子注入(具有10-3cm2/vs的体电荷载流子迁移率)的理想化欧姆接触,这在先前为不可能的。
本发明可提供至具有高达5.8ev的ip的半导体的欧姆接触。该方法适用于在场效晶体管中进行至具有由4.5ev至5.8ev的ip的有机半导体,尤其至具有由5.0ev至5.8ev的ip的半导体的欧姆接触,这特别具有挑战性。
本发明还可通过简单扩展而应用于基于金属氧化物、有机晶体、有机-无机混合晶体、无机纳米晶体与纳米线、富勒烯、碳与其他纳米管和石墨烯作为半导体的其他类型的场效晶体管。
图1显示出并入在其与结合阴离子层(分别为16、26、36、46)的界面(分别为17、27、37、47)处p掺杂的半导体层(分别为12、22、32、42)的四个可能fet器件构造的示意性表示:(a)底接触底栅极晶体管构造,(b)顶栅极底接触晶体管构造,(c)底栅极顶接触晶体管构造,(d)顶栅极顶接触晶体管构造。“底栅极”构造是指其中栅极层(15、25、35、45)和栅极介电层(14、24、34、44)在半导体层(12、22、32、42)下面的那些构造。“顶栅极”构造是指其中栅极层和栅极介电层在半导体层上面的那些构造。“底接触”构造是指其中源极电极(11、21、31、41)和漏极电极(13、23、33、43)层在半导体层下面的那些构造。“顶接触”构造是指其中源极和漏极电极层在半导体层上面的那些构造。
图2显示出在与结合阴离子层的界面处并入p掺杂半导体的bgbc构造的制备方法。
图3显示出来自au上的原始3-nm厚dppt-t2有机半导体膜覆盖层(长黑虚线)与由au上的氧化剂阳离子pedt:psstpam抗衡的结合阴离子层(黑色实线)的差示光谱的p掺杂dppt-t2有机半导体膜(短黑色虚线)的拉曼光谱学证据。
图4显示出超薄p掺杂dppt-t2有机半导体的紫外线光电子光谱学证据。20-nm厚dppt-t2原始膜在au上(最左边面板)。沉积在由au上的旁观阳离子(psscs)抗衡的结合阴离子层上的3-nm厚dppt-t2膜的ef-homo能隙减小(中间面板)且在沉积在由au上的氧化剂阳离子(psstpa)抗衡的结合阴离子层上时ef-homo能隙进一步减小(右面板)。
图5显示出由7-nmcr/30-nmau上的旁观阳离子(psscs)抗衡的结合阴离子分子薄层的x射线光电子光谱学证据。(a)s2p、cs4p(b)cs3d(c)cl2p
图6显示出由7-nmcr/30-nmau上的氧化剂阳离子(psstpa)抗衡的结合阴离子的分子薄层的x射线光电子光谱学证据(a)s2p、cs4p(b)cs3d(c)cl2p。
图7显示出掺杂的dppt-t2有机半导体的x射线光电子光谱学证据。(a)7-nmcr/30-nmau上的20-nm厚原始dppt-t2有机半导体膜的s2p核级xps;(b)由氧化剂阳离子(psstpa)抗衡的结合阴离子的分子薄层上的3-nm厚原始dppt-t2有机半导体膜的s2p核级xps(c)(a)与(b)的差示光谱显示向高结合能尾的扩展。
图8显示出在改性和不改性au源极-漏极电极的情况下dppt-t2有机半导体顶栅极底接触场效晶体管的转移(左面板)和输出(右面板)曲线(a)au(b)psscs/au(c)pssh/au(d)psstpa/au(e)pssno/au。
图9显示出对使用由氧化剂阳离子(psstpa)抗衡的结合阴离子层的改进dppt-t2有机半导体顶栅极底接触场效晶体管使用传输线方法(tlm)在一定范围的标准化电流(i)和栅极电压(vg)值内提取的接触电阻(rc)值的分散。
实施例
实施例1:结合阴离子层的中间分子薄层的制备
实施例1a:聚(苯乙烯磺酸)(pssh)-离子交换的
使用聚(苯乙烯磺酸)(pssh,科学聚合物产品公司,mw70k)。通过重量分析测定其浓度为200mg/ml。将1g阳离子交换树脂(
实施例1b:聚(苯乙烯磺酸)(pssh)-渗析的
使用聚(苯乙烯磺酸)(pssh,科学聚合物产品公司,mw70k)。通过重量分析测定其浓度为200mg/ml。将其在0.1mhcl溶液中渗析且使其搅拌2h,重复三次且最终用
实施例1c:聚(苯乙烯磺酸铯)(psscs)-离子交换的
使用聚(苯乙烯磺酸)(pssh,科学聚合物产品公司,mw70k,200mg/ml)。将1g阳离子交换树脂(
实施例1d:聚(苯乙烯磺酸铯)(psscs)聚阴离子中间层-渗析的
使用聚(苯乙烯磺酸)(pssh,科学聚合物产品公司,mw70k,200mg/ml)。将其在0.1mcscl溶液中渗析且使其搅拌和旋转2h,重复三次且最终用
实施例1e:聚(苯乙烯磺酸锂)(pssli)-离子交换的
使用聚(苯乙烯磺酸)(pssh,科学聚合物产品公司,mw70k,200mg/ml)。将1g阳离子交换树脂(
使用所购买的聚(苯乙烯磺酸钠)(pssna,fluka)。将其溶解至
实施例1g:聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸)(pedt:pssh)-渗析的
使用所购买的聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸)(pedt:pssh,heraeuspreciousmetalsgmbh&co,比率1:6)。将其在0.1mhcl溶液中渗析且使其搅拌2h,重复三次且最终用
实施例1h:聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸铯)(pedt:psscs)-渗析的
使用所购买的聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸)(pedt:pssh,heraeuspreciousmetalsgmbh&co,比率1:6)。将其在0.1mcscl溶液中渗析且使其搅拌和旋转2h,重复三次且最终用
实施例2:氧化剂溶液的制备
实施例2a:六氯锑酸三(4-溴苯基铵)(tpa+sbcl6-)
使用所购买的六氯锑酸三(4-溴苯基铵)(tpa+sbcl6-,sigma-aldrich)。制备无水碳酸亚丙酯中的30mm的tpam+sbcl6-(sigma-aldrich)。作为替换,可在无水硝基甲烷中或在无水乙腈中或在1:1无水硝基甲烷:甲苯中制备30mmtpamsbcl6溶液。
实施例2b:六氟锑酸亚硝
使用所购买的六氟锑酸亚硝
实施例2c:六氟锑酸噻蒽
使用所购买的噻蒽。单独地,制备无水三氯甲烷中的0.25m噻蒽和无水硝基甲烷中的0.5mno+sbf6-。然后以1.1:1.0的化学计量比将0.19ml六氟锑酸亚硝
实施例3:改进fet效能
实施例3a:由与六氯锑酸三(4-溴苯基铵)(tpa+sbcl6-)离子交换的聚(苯乙烯磺酸铯)(psscs)制备具有氧化剂的结合阴离子的分子薄层
通过氧等离子体清洁用具有l=10、20、50、100微米和通道长度1厘米的可变通道长度的7-nmcr/30-nmau源极-漏极电极制备的0.2-mm厚聚对苯二甲酸乙二醇酯(pet)基材(24秒,270w)。然后在60℃下将氧等离子体清洁的基材浸没于2mg/mlpsscs溶液中10分钟以在au源极-漏极电极上沉积psscs膜的超薄层。然后以5000rpm将该基材旋转干燥且将其退火至120℃以进一步促进au电极上的超薄层的粘合。用
图4显示出超薄p掺杂dppt-t2有机半导体的紫外线光电子光谱学证据。
图6显示出由7-nmcr/30-nmau上的氧化剂阳离子(psstpa)抗衡的结合阴离子的分子薄层的x射线光电子光谱学证据。
图7显示出掺杂的dppt-t2有机半导体的x射线光电子光谱学证据。
图8显示出在改性和不改性au源极-漏极电极的情况下dppt-t2有机半导体顶栅极底接触场效晶体管的转移(左面板)和输出(右面板)曲线。
实施例3b:由氧化剂阳离子抗衡的结合阴离子的分子薄层为pssno
如实施例3a中所述而制备fet,其中由用无水碳酸亚丙酯中的no+sbf6-的30mm氧化剂溶液处理的0.5-2nmpsscs膜制备结合阴离子的分子薄层。使用经改性tlm提取的该fet的接触电阻为22kωcm。
图8显示出在改性和不改性au源极-漏极电极的情况下dppt-t2有机半导体顶栅极底接触场效晶体管的转移(左面板)和输出(右面板)曲线。
实施例3c:由氧化剂阳离子抗衡的结合阴离子的分子薄层为pssthia
如实施例3a中所述而制备fet,其中结合阴离子的分子薄层为用在无水碳酸亚丙酯中溶解的thia+sbf6-的30mm氧化剂溶液处理的3nmpsscs膜。
实施例3d:由氧化剂阳离子抗衡的结合阴离子的分子薄层为pedt:psstpa。
如实施例3a中所述而制备fet,其中由用在无水碳酸亚丙酯中溶解的tpa+sbf6-的30mm氧化剂溶液处理的2-3nmpedt:pssh膜制备结合阴离子的分子薄层。
实施例3e:由氧化剂阳离子抗衡的结合阴离子的分子薄层为pedt:pssthia
如实施例3a中所述而制备fet,其中由与在无水碳酸亚丙酯中溶解的thia+sbf6-的30mm氧化剂溶液离子交换的2-3nmpedt:psscs制备结合阴离子的分子薄层。
实施例3f:由氧化剂阳离子抗衡的结合阴离子的分子薄层为pedt:psstpa
如实施例3a中所述而制备fet,其中由与无水碳酸亚丙酯中的tpa+sbcl6-的30mm氧化剂溶液离子交换的2-3nmpedt:psscs制备结合阴离子的分子薄层。
图3显示出p掺杂dppt-t2有机半导体膜的拉曼光谱学证据。
实施例4:较低fet效能
实施例4a:结合阴离子的分子薄层为聚(苯乙烯磺酸铯)(psscs)。
通过氧等离子体清洁用具有l=10、20、50、100微米和通道长度1厘米的可变通道长度的7-nmcr/30-nmau源极-漏极电极制备的0.2-mm厚聚对苯二甲酸乙二醇酯(pet)基材(24秒,270w)。然后在60℃下将氧等离子体清洁的基材浸没于2mg/mlpsscs溶液中10分钟以在au源极-漏极电极上沉积psscs膜的超薄层。然后以5000rpm使该基材旋转干燥且将其退火至120℃以进一步促进au电极上的超薄层的粘合。用
图5显示出由7-nmcr/30-nmau上的旁观阳离子(psscs)抗衡的结合阴离子分子薄层的x射线光电子光谱学证据。
图8显示出在改性和不改性au源极-漏极电极的情况下dppt-t2有机半导体顶栅极底接触场效晶体管的转移(左面板)和输出(右面板)曲线。
- 还没有人留言评论。精彩留言会获得点赞!