68Ge/68Ga发生器的制作方法
本发明涉及与权利要求1的前序部分一致的用于连续生产68ga子核素的锗-68/镓-68(68ge/68ga)发生器。本发明还涉及根据权利要求16的选自钒、铌和钽的金属的至少一种氧化物的用途。68ga是具有高度吸引力的发射正电子的放射性核素,其同时在正电子发射断层扫描(pet)的使用中发挥重要作用。对于68ga在诊断学中的用途、特别是它与dotatoc的螯合的简介和概述,参考申请人的ep2439747b1。
背景技术:
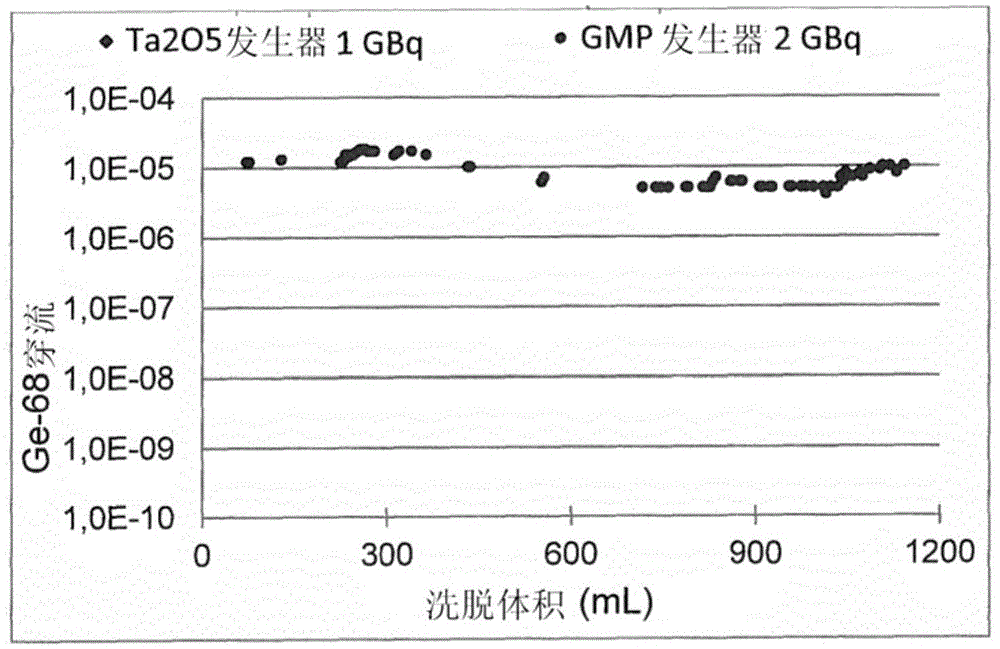
:在提供纯的放射性核素中,元素特异性吸附剂极为令人感兴趣。对于化学科学的不同领域来说,也是如此。然而,特别令人感兴趣的是将这些材料应用于分析和医学用途的放射性核素的分离和生产。锗特异性材料的制药应用的关键实例是68ge/68ga放射性核素发生器系统。这种放射性核素发生器描述在例如ep2216789a1中。所述68ge/68ga放射性核素发生器是基于放射性核素68ge在锗特异性材料上的吸附。相对长寿命的68ge(t1/2=270.82d)产生短寿命的中间体同位素68ga(t1/2=67.6min)。当68ge被固定在支持材料上时,连续形成的68ga可以被重复地洗脱(生产)。68ga是一种正电子发射体(β+分支=89%),其可用于通过配位标记制备放射性药物。在过去的几年期间,使用68ga标记的dota偶联的肽的肿瘤成像已成为使用pet和pet/ct诊断神经内分泌和其他肿瘤和转移肿瘤的已确立的方法。68ga在医学用途中的关键优点(成本、后勤优点)是它通过68ge/68ga放射性核素发生器的可获得性[1,2]。现有技术的68ge/68ga放射性核素发生器的基本质量参数是68ge的不想要的穿流(breakthrough)、68ga的洗脱得率和洗脱稳定性。68ge穿流的限度与度量68ga洗脱物的药品质量的其他参数一起,提供在《欧洲药典》专著(europeanpharmacopoeiamonograph)中[3]。68ga的洗脱得率和洗脱稳定性是68ge/68ga发生器的效率和储存期限的重要因素。为了用68ga对药品进行成功的放射性标记,所使用的68ga制备物必须满足对化学品和放射化学品质量的高要求。68ga必须以其“离子”形式生产(即没有任何络合剂)。68ga制备物只能以小体积并在低酸度下用于配位标记。所述制备物必须不含作为镓在生物分子中的掺入的强烈竞争剂的金属杂质[4-6]。当前在全球市场上可获得的68ge/68ga放射性核素发生器系统是基于使用无机离子交换剂或有机分子作为吸附剂(表1)。最常用的无机离子交换剂是tio2(cyclotroncompany,russianfederation,和eckert&zieglerisotopeproducts,germany)和sno2(ithembalabs,southafrica)。这些发生器类型的特征是68ga制备物被来自于所使用的支持材料的痕量其他金属污染和对洗脱剂的高酸度和/或大体积的要求。因此,为了利用现有技术可获得的基于金属氧化物的放射性核素发生器制备68ga标记的放射性药物,对获得的68ga洗脱物的预处理是必需的[5-8]。无机离子交换剂的可替选方案是有机聚合物,其引入有带有对锗具有高亲和力的官能团的单一分子,如ep2439747b1中所述。这些分子可以是焦没食子酸、儿茶酚等,其通过酚羟基与锗形成强络合物。关键实例是市场上唯一的无金属68ge/68ga放射性核素发生器系统(itgisotopetechnologiesgarchinggmbh,germany),其基于使用焦没食子酸衍生的sio2作为吸附剂[7,9](表1)。这种发生器详细描述在申请人的ep2439747b1中。这种现有技术的68ge/68ga放射性核素发生器已经允许对生物分子进行高效的放射性标记而不需要68ga洗脱物的预先纯化。然而,在放射性核素发生器系统中使用的基于有机物的吸附剂在暴露于高剂量辐射时是辐射分解不稳定的。因此,吸附剂的改进的化学稳定性在发展用于在更高的68ge活性下获得改进的性能的68ge/68ga发生器中发挥重要作用。一般来说,与68ge/68ga发生器的吸附剂的性质相关的一些因素影响68ga洗脱物的关键质量参数。吸附剂的低的化学稳定性在高辐射分解应力条件下增加68ge的穿流。此外,在发生器的储存期限内,68ge活性区通过洗脱沿着吸附剂柱移动,使锗倾向于在金属氧化物的晶格缺陷或焦没食子酸衍生物的碳链和二氧化硅的网络内部分扩散。这些扩散现象可能是引起对于市场上的68ge/68ga发生器来说常见的通过洗脱获得的68ga的洗脱得率降低的因素。考虑到ep2439747b1的现有技术,本发明的目的是提供一种改进的68ge/68ga放射性核素发生器,其在68ga洗脱期间显示出可忽略的68ge穿流,对辐射分解稳定,特别是在涉及较高的68ge活性时,并同时提供高的68ga得率,并且最后,基本上没有不想要的杂质。这个目的通过根据权利要求1的68ge/68ga发生器和根据权利要求16的选自钒、铌和钽的金属的氧化物作为无机支持材料用于制造68ge/68ga发生器的用途来实现。技术实现要素:具体来说,本发明涉及:一种用于连续生产68ga子核素的68ge/68ga发生器,其中其68ge母核素被特异性吸附到无机支持材料,并且其中所述68ge母核素以270.82d的半衰期通过电子俘获连续衰变成68ga,其中所述无机支持材料是选自钒、铌和钽的金属的至少一种氧化物。本发明还涉及选自钒、铌和钽的金属的至少一种氧化物的用途,其作为无机支持材料用于制造符合本发明的用于连续生产68ga子核素的68ge/68ga发生器,其中其68ge母核素被特异性吸附到无机支持材料,并且其中所述68ge母核素以270.82d的半衰期通过电子俘获连续衰变成68ga。本发明的优选实施方式是一种68ge/68ga发生器,其中所述氧化物是具有通式(1)的氧化物:m2o5(1),其中m表示钒、铌或钽。在本发明中使用的特别优选的氧化物是五氧化二钽(ta2o5),其可以以其α-和/或β-晶型使用。在本发明中用作支持材料的氧化物可以通过下述步骤来获得:水解通式(2)的金属卤化物:mx5(2),其中m表示钒、铌或钽;并且x表示氯、溴或碘;并将从所述水解得到的金属氢氧化物通过退火转变成所需的金属氧化物。本发明的优选实施方式是使用tacl5作为所述金属卤化物,其水解产生ta(oh)5。或者,根据本发明,所需氧化物也可以通过将金属粉末在氧气气氛下退火来获得,其中所述金属选自钒、铌和钽,其中钽优选作为所述金属,并且所得到的氧化物是ta2o5。在本发明的另一个优选实施方式中,所述氧化物的粒度分布为5μm至300μm,特别是10μm至200μm。通常,所述68ge母核素以68ge(iv)阳离子、特别是68ge水合阳离子的形式被吸附到所述氧化物支持材料,所述两种阳离子都可以容易地获得。根据本发明的另一个优选实施方式,用0,01至0,1mhcl、特别是用0,05mhcl从所述68ge/68ga发生器洗脱所述68ga。在本发明的另一个优选实施方式中,68ge的穿流率在1000mbq的初始活性下<10-3%,特别是<10-6%,优选地<10-7%,并且在2000mbq的初始活性下<4x10-7%。这远低于欧洲药典所要求的值[11],并且远低于市场上可获得的任何68ge/68ga发生器(参见下面的表1)。68ga的典型洗脱得率大于65%。本发明涉及使用新的锗特异性吸附剂,即属于其中金属可以是钒、铌和钽的金属氧化物类别的氧化物。也可以使用混合金属氧化物或不同氧化物的混合物。具体来说,五氧化物已被证明对总体来说ge、具体来说68ge是适合的吸附剂。尽管来自于上述钒族金属的所有氧化物通常都起到特异性锗吸附剂的作用,但在实践中已证明五氧化二钽(ta2o5)是最优选的。符合本发明的吸附剂可以从其相应的五氯化物例如五氯化钽通过水解途径,或从v、nb或ta或其混合物的金属粉末通过退火途径来合成,其中优选地使用钽粉末。所述金属粉末的氧化在常压大气条件下进行。符合本发明的吸附剂的制药用途允许通过新的68ge/68ga放射性核素发生器提高发射正电子的医用放射性核素68ga的生产可能性。所述吸附剂的化学本质能够实现68ge的高效吸附,68ga的高效且稳定的解吸,68ge的非常低的穿流和生物分子用68ga的高效标记。与基于现有技术的其他金属氧化物吸附剂例如tio2或sno2的当前系统相比,所需的放射性核素68ga可以以高的化学和放射化学纯度直接生产(即没有任何预处理),用于制备68ga标记的放射性药物。此外,所述吸附剂是化学惰性和对辐射分解稳定的,这允许它被成功地用于具有高性能的高活性的放射性核素发生器中。到目前为止,没有关于将元素周期表的钒族金属氧化物、特别是五氧化二钽作为吸附剂用于68ge/68ga类型的放射性药物放射性核素发生器中的文献。此外,通过下文公开的方法合成符合本发明的金属氧化物例如五氧化二钽吸附剂,适合于本发明的目的。所获得的吸附剂通过不同的固态技术例如x-射线衍射、扫描电子显微术、傅里叶变换红外光谱术和通过brunauer-emmet-teller方法的表面积测量进行了充分表征。这种全面分析产生了良好表征的吸附剂,其具有优化的关键参数68ga的洗脱性能、68ge的穿流、吸附剂的容量和标记性质。下面的表1给出了目前在市场上可以获得的68ge/68ga发生器系统的概述。表1的最后一行显示了符合本发明的68ge/68ga发生器。表1.目前市场上可以获得的68ge/68ga发生器的技术规格。表1中的信息部分获取自roesch2015[10]。*1由cyclotronco.ltd,obninsk,russia提供*2在200次洗脱后*3表示成洗脱物中8ge/68ga的放射活性的比率*4在300天后*5仅适用于每日洗脱的值*6在校准当天,在4ml0.05mhcl中*7在校准时68ga中的68ge含量发明详述本发明涉及选自钒氧化物、铌氧化物和钽氧化物、特别是五氧化二钽(ta2o5)的新的锗特异性吸附剂在68ge/68ga放射性核素发生器中的用途。所述化学惰性且稳定的吸附剂能够实现68ge的高效吸附,68ga的高效且稳定的解吸,68ge的非常低的穿流和生物分子用68ga的高效标记。附图说明从下面的实施例描述以及附图,本发明的其他特点和优点将变得明显:图1是两种68ge/68ga发生器的68ge穿流百分率随累积洗脱体积的变化的图形表述。符合ep2439747b1的当前itggmp发生器与符合本发明的基于ta2o5的发生器之间的比较显示出68ge穿流水平的显著差异;图2是两种68ge/68ga发生器的68ga洗脱得率随累积洗脱体积的变化的图形表述。符合ep2439747b1的当前itggmp发生器与符合本发明的基于ta2o5的发生器之间的比较显示出68ga洗脱得率的稳定性的巨大差异;图3示出了hplc色谱图,其呈现了在1900mbq68ge/68ga发生器的90h的向内生长时间后来自于使用68ga的直接标记的结果。游离的未标记的68ga和标记的68ga的百分率分别为1.73%和96.42%;图4a和图4b是呈现了β-ta2o5(图4a)和α-ta2o5(图4b)的表面结构的扫描电子显微镜(sem)图像的图像。从所述图像可以看到不同的表面特征,指示了β-ta2o5的更高的表面积(图4a),表明当用于68ge/68ga发生器柱中时更高的容量;图5示出了使用ta2o5吸附剂的68ge/68ga发生器的68ga的洗脱曲线。发生器的初始68ge活性是1000mbq;图6示出了68ga的洗脱得率随洗脱体积变化的图,显示出结果大于70%(3000mbq);图7示出了68ge穿流的图,其中的值低于10-7%;以及图8示出了使用ta2o5吸附剂的68ge/68ga发生器的68ga的洗脱曲线。发生器的初始68ge活性是4000mbq。具体实施方式实施例1:高达1000mbq(27mci)68ge的发生器以符合本发明的最优选的金属氧化物五氧化二钽的制造为例描述了下述合成方法。然而,本领域普通技术人员将会理解,该合成方法可以被容易地用于制造本发明的其他优选实施方式即五氧化二钒和五氧化二铌,特别是由于它们相近的化学性质。ta2o5的合成用于ta2o5吸附剂的合成法由申请人开发,使用了两种主要合成途径:使用五氯化钽(tacl5)作为起始原料的水解途径,和使用钽粉(ta粉)作为起始原料的退火途径。水解途径tacl5的水解在水中使用受控的水/tacl5比率进行。在水解过程中调节水的温度并保持稳定,以便控制终产物ta2o5的粒度。在固相研究的基础上选择氢氧化钽(ta(oh)5)的退火温度,以便找出在68ge/68ga放射性核素发生器中使用的吸附剂的最佳性能。退火途径ta粉的氧化使用具有选定粒度分布的起始原料来进行。在固相研究的基础上选择ta粉的退火温度,以便找出在68ge/68ga放射性核素发生器中使用的吸附剂的最佳性能。合成的ta2o5的技术规格在放射性药物68ge/68ga发生器中使用的合成的ta2o5的技术规格包括下述标准:退火温度,粒度分布,68ge与吸附剂之间的分配系数(kd)和68ga的解吸(可洗脱性)。所述标准概述在下面的表2中。表2.合成的ta2o5的技术规格。技术规格标准退火温度600-1350℃分配系数(kd)2000-20000ml/g68ga的可洗脱性≥65%粒径分布10-200μmta2o5吸附剂的表征在五氧化二钽吸附剂的合成的开发期间,研究了分别与68ge和68ga的吸附和解吸性质相关的不同参数(表3)。这些参数包括ta2o5的晶体结构和表面形态、表面积和粒度分布。将通过放射化学分析为68ge(分配系数(kd)和容量)和68ga(可洗脱性)获得的结果,通过经分析技术获得的观察和结果进行关联,所述分析技术例如用于晶体结构分析的x-射线衍射(xrd)、用于表面形态研究的扫描电子显微术(sem)(图4)、表面积测定(brunauer-emmet-teller(bet))和粒度分布的测定。表3.合成的ta2o5吸附剂材料的不同技术规格之间的关联性*1分离的直径<8μm的粒子在与发生器相关的溶液ph(0.05mhcl)和ge浓度([ge总]<0.005m)下,四价锗以锗酸(ge(oh)4)的形式存在[12,13]。在这些条件下,锗与五氧化二钽表面上的羟基结合[14]。实验已表明68ge的小的粒度和大的表面积与高效吸附之间的明显的正相关性。另一方面,小的粒度对68ga的可洗脱性的效率具有负面影响。这就是开发所述ta2o5的合成方法的主要目标是最小化小粒子(<10μm)的形成和提高ta2o5粒子的表面积的原因。在图4中,可以看到ta2o5的两种晶型的表面差异:由β-ta2o5形成的粒子(图4a)具有被在水解的攻击性化学条件期间形成的凹陷和构造所覆盖的表面;因此与α-ta2o5的粒子(图4b)相比提供了更大的表面积和对68ge更高的kd和容量,α-ta2o5粒子的这些形态结构已由于高的退火温度而“熔化”。另一方面,α-ta2o5产生的光滑的表面特征为68ga提供了更好的可洗脱性质。结论:目的是开发一种合成ta2o5吸附剂的方法,所述ta2o5吸附剂具有68ge的高效吸附(储存期限)与68ga的高效可洗脱性(洗脱得率)之间的理想平衡。遵照下述水解途径合成了一批锗特异性吸附剂:将五氯化钽(tacl5)与热水(80℃,固体/液体比率为20g/l)混合以产生氢氧化钽(ta(oh)5),将其在900℃下退火超过24h,以便形成结晶钽氧化物(ta2o5)。在分离粒度范围为10μm–200μm的粒子后,将最终材料作为吸附剂用于68ge/68ga发生器。向两个发生器柱装填已知量的所述吸附剂(8g)。将所述柱装载已知量的68ge(1000mbq、2000mbq)和稳定的ge(ge的总质量=80μg)。所述放射性核素发生器在gmp条件下生产。使所述68ge/68ga发生器经历洗脱程序,关键参数如下。在当前的洗脱程序阶段,下述与关键参数相关的值是有效的:-当前的总累计洗脱体积:700ml(1000mbq),400ml(2000mbq)-68ga的洗脱得率:>65%,稳定-当前:70%(1000mbq),73%(2000mbq)(图2)-洗脱体积:6ml(图6)-68ge的穿流:<10-6%(ph.eur.的水平:10-3%)-当前:10-7%(1000mbq),4x10-7%(2000mbq)(图1)-标记效率:在90h的向内生长期后通过直接洗脱(68ga-dota-toc)>96%(图3)。关键质量参数:68ge的穿流和68ga的洗脱得率一般来说,与68ge/68ga发生器的吸附剂的性质相关的一些因素影响68ga洗脱物的关键质量参数。吸附剂的低的化学稳定性在高辐射分解应力条件下增加68ge的穿流。此外,在发生器的储存期限内,68ge活性区通过洗脱沿着吸附剂柱移动,使锗倾向于在金属氧化物的晶格缺陷或焦没食子酸衍生物的碳链和二氧化硅的网络内部分扩散。这些扩散现象可能是引起对于市场上的现有技术68ge/68ga发生器来说常见的通过洗脱获得的68ga的洗脱得率降低的因素。最初选择五氧化二钽用作吸附剂是出于两个原因:它是化学惰性且稳定的材料,这使它适合于在高辐射分解的条件下使用并令人吃惊地获得低的68ge穿流(图1)。此外,钽阳离子(主要)处于五价氧化态这一事实被视为对68ga洗脱得率的稳定性有益,并防止四价锗扩散到ta2o5的晶格中(图2)。这些性质、即化学稳定性和本质的证据,可以在分别呈现了gmp发生器和ta2o5发生器中68ge的穿流和68ga的洗脱得率的行为随着累积洗脱体积的变化的图1和2中看到。关键质量参数:发生器的标记性质使用合成的ta2o5吸附剂的放射性药物68ge/68ga发生器的标记性质,通过基于欧洲药典专著的方法来试验[11]。对从标称68ge活性为1900mbq并且向内生长时间(无洗脱时间)为90小时的发生器洗脱的68ga洗脱物进行了所述试验。所述试验的目的是证实即使在较长时间段的无洗脱期间ta2o5吸附剂针对辐射分解的稳定性。通过高压液相色谱(hplc)从直接标记获得的结果是标记产物的得率超过96%。这清楚地证实了在68ge/68ga发生器中使用的ta2o5吸附剂的广泛稳定性,并且表明为了获得用于放射标记的功能完全的发生器,不需要在周末后进行清洗(图3)。实施例2:超过1850mbq(50mci)68ge的发生器向发生器柱装填已知量的所述吸附剂(8-9g)。将所述柱装载已知量的68ge(4000mbq)并且不添加稳定ge(ge的总量通过ge-68的比活性计算为44μg)。使所述68ge/68ga发生器经历洗脱程序,关键参数如下。在当前的洗脱程序阶段,下述与关键参数相关的值被示出在图6、7和8中:具体来说,图6示出了其中将以%为单位的洗脱得率值对以ml为单位的洗脱体积作图的图,显示出68ga的洗脱得率超过70%(3000mbq)。图7示出了68ge的穿流值小于10-7%。最后,图8示出了使用ta2o5吸附剂的68ge/68ga发生器的68ga的洗脱曲线。所述发生器的初始68ge活性为4000mbq。所述发生器显示出>3000mbq的68ga得率。使用常用68gapet示踪剂例如psma-11(hbed-cc)和dotatate的典型的标记程序显示出在生产结束时(生产结束通常是发生器洗脱后30min至60min)55mci([ga-68]ga-hbed-cc)和45mci([ga-68]ga-dotatate)的结果。基于100gbq/mg68ge的比活性和8-9gta2o5发生器柱上标称的总可用锗量的计算,发生器可以装载8000mbq68ge(对应于80μg锗)。当将ta2o5用它的对应氧化物nb2o5和v2o5替换时,可以获得与实施例1和2中指明的相似的洗脱曲线、穿流值、得率(数据未示出)。它们的合成基本上遵照与上面实施例1中描述的相同的途径。使用本发明,可以满足对数量充足且质量可靠的放射性药物级68ga的临床需求。参考文献[1]al-nahhasa,winz,szyskot,singhaa,nanniic,fantis,rubellod.镓-68pet:受体癌症成像中的新前沿(gallium-68pet:anewfrontierinreceptorcancerimaging),anticancerres.2007;27:4087-4094.[2]maeckeh,hofmannm,haberkornu.肿瘤成像中的68ga标记的肽(68ga-labeledpeptidesintumorimaging),jnucmed.2005;46:172s-178s.[3]用于放射性标记的氯化镓(68ga)溶液(gallium(68ga)chloridesolutionforradiolabelling),ph.eur.7.8.07/2013:2464;5643-5644.[4]breemanw,jongm,bloise,bernardb,konijnenbergm,krenninge.用68ga对dota-肽进行放射标记(radiolabellingdota-peptideswith68ga),eurjnucmedmolimaging.2005;32:478-485.[5]meyerg-j,h,schuhmacherj,knappw,hofmannm.68ga标记的dota衍生的肽配体(68ga-labelleddota-derivatisedpeptideligands),eurjnucmedmolimaging.2004;31:1097-1104.[6]zhernosekovk,filosofovd,baumr,aschoffp,bihlh,razbasha,jahnm,jenneweinm,f.用于医学应用的发生器生产的68ga的加工(processingofgenerator-produced68gaformedicalapplication),jnucmed.2007;48:1741-1748.[7]zhernosekovk,nikulat.用于功能化支持物的分子、放射性核素向所述支持物的附连和用于制备所述放射性核素的放射性核素发生器以及制备方法(moleculeforfunctionalizingasupport,attachmentofaradionuclidetothesupportandradionuclidegeneratorforpreparingtheradionuclide,andpreparationprocess),patentus2010/0202915a1.[8]chakravartyr,chakrabortys,ramr,vatsar,bhusarip,shuklaj,mittalbr,dasha.不同68ge/68ga发生器的详细评估:对获得高效68ga放射性药物的尝试(detailedevaluationofdifferent68ge/68gagenerators:anattempttowardachievingefficient68garadiopharmacy),jlabelcompdradiopharm.2016;59:87-94.[9]schuhmacherj,maier-borstw.用于在稀hcl中生产68ga的新的68ge/68ga放射性同位素发生器系统(anew68ge/68garadioisotopegeneratorsystemforproductionof68gaindilutehcl),japplradisotopes.1981;32:31-36.[10]roeschf.68ga放射性药物:当前状态和未来(68garadiopharmaceuticals:currentstatusandfuture),in:internationalconferenceonclinicalpet-ctandmolecularimaging(ipet2015).vienna,austria,5-9october2015.[11]镓(68ga)依多曲肽注射剂(gallium(68ga)edotreotideinjection),pa/ph/exp.14/t(07)12anp;e1-e7.[12]baescf,mesmerre.阳离子的水解(thehydrolysisofcations),robertekriegerpublishingcompany,malabar,florida,usa.1986.[13]woodsa,samsonim.镓、锗、铟和钪的水文地球化学(theaqueousgeochemistryofgallium,germanium,indiumandscandium),oregeolrev.2006;28:57-102.[14]pokrovskyos,pokrovskigs,schottj,galya.锗在针铁石上的吸附和锗与氢氧化铁的共沉淀的实验研究:x-射线吸附精细结构和宏观表征(experimentalstudyofgermaniumadsorptionongoethiteandgermaniumcoprecipitationwithironhydroxide:x-rayabsorptionfinestructureandmacroscopiccharacterization),geochimcosmochimac.2006;70:3325-3341.当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1