蛋白质的选择性还原的制作方法
本申请要求2014年2月11日提交的美国专利申请号61/938,378的优先权,其内容通过引用全文纳入本文。
发明背景
选定的氨基酸被突变成半胱氨酸Cys的单克隆抗体(例如工程化的半胱氨酸mAbs,即ecmAbs)特别适合用于偶联物(例如抗体药物偶联物(ADCs)),因为这些由其衍生的偶联物具有有利的特性,其中包括同质性、有利的药物动力学、稳定性和溶解性。这些半胱氨酸突变被置于在抗体氨基酸序列中的某些位置,这些位置通常不会形成链间或者链内的二硫键,并且在细胞内产生突变mAb的表达机制会将半胱氨酸残基作为未配对的半胱氨酸进行处理。因此,工程化半胱氨酸,通常会用非编码的半胱氨酸分子以混合的二硫键产物形式表达(例如,工程化半胱氨酸通常用封端剂(例如半胱氨酸、半胱氨酰甘氨酸、谷胱甘肽)进行“封端(capped)”)。
因此,为了从ecmAbs出发制备偶联物(例如ADC),通常需要将ecmAb置于还原条件下,从而将工程化半胱氨酸从混合的二硫键产物转化为自由巯基,通常这一“脱封端”或者“活化”伴随着ecmAb链间二硫键的还原。尽管链间二硫键可以通过温和氧化重新形成,但是这种再氧化步骤增加了ADC制备过程中复杂性和费用。选择性还原方法是难以实现的,因为已证明难以还原工程化半胱氨酸残基的同时而不还原链间二硫键。因此,需要开发用于对工程化半胱氨酸残基的进行脱封端的选择性还原方法。本发明解决了这个和其它需求。
发明概述
在本发明中,提供了一种选择性还原工程化的半胱氨酸抗体的方法。这种方法包括:将一还原剂与工程化的半胱氨酸抗体分子接触,每个抗体分子含有至少一个封端的工程化的半胱氨酸残基和至少一个链间二硫键,以及将还原剂与抗体分子在足以使工程化的半胱氨酸残基“脱封端”并且形成封端副产物的条件下进行反应。封端副产物在还原反应的过程中被移去。这种方法导致形成脱封端的工程化半胱氨酸抗体制剂。这种脱封端的工程化半胱氨酸抗体制剂可以包含封端的和脱封端的工程化半胱氨酸抗体。还原反应前存在于抗体分子中基本上所有的链间二硫键,在脱封端的工程化半胱氨酸抗体制剂中仍然保留。所述方法可以进一步包括以下步骤:在移除封端副产物时,向还原反应补充额外的还原剂。在还原反应过程中从反应混合物中移除封端副产物,可以通过例如透析或渗滤进行。在优选的实施方式中,在还原反应混合物中,封端副产物浓度被维持低于如下浓度:在该浓度下,再封端阻止了工程化半胱氨酸残基被进一步活化。
在某些方面,所述方法提供一种非封端的工程化半胱氨酸抗体制剂。典型地,在抗体制剂中至少60%的工程化的半胱氨酸是非封端工程化的半胱氨酸残基。在某些方面,在抗体制剂中至少70%,至少75%或者至少80%的工程化的半胱氨酸残基是非封端工程化的半胱氨酸残基。在某些方面,采用本发明方法,在抗体分子中至少85%的还原反应前就存在的链间二硫键在脱封端工程化的半胱氨酸抗体制剂中仍然保留。在某些方面,选择还原剂和还原条件,使得不超过20%,不超过15%,不超过10%或者不超过5%的抗体分子中存在的链间二硫键,在还原反应过程中被转化成一对自由巯基。
在相关方面,这个发明提供抗体偶联物,包括抗体药物偶联物,和采用脱封端抗体制备抗体偶联物的方法。在制备抗体药物偶联物之前,抗体制剂中残留的还原剂被移除。
本发明还提供一种选择性还原含有非配对半胱氨酸残基的非抗体蛋白质的方法。这种方法包括:将一还原剂与蛋白质分子接触,每个蛋白质分子含有至少一个封端的工程化的半胱氨酸残基和至少一个链间二硫键,以及将还原剂与蛋白质分子在足以使半胱氨酸残基“脱封端”并形成封端副产物的条件下进行反应。封端副产物在还原反应的过程中被移去。所述方法导致形成脱封端的半胱氨酸蛋白质制剂。这种脱封端的半胱氨酸蛋白质制剂可以包含封端和脱封端的半胱氨酸残基的蛋白质分子。蛋白质分子中在还原反应前就存在的基本上所有的链间二硫键,在脱封端的半胱氨酸蛋白质制剂中仍然保留。这种方法可以进一步包括以下步骤:在移除封端副产物时,向还原反应补充额外的还原剂。在还原反应过程中从反应混合物中移除封端副产物可以通过例如透析或渗滤进行。在优选的实施方式中,在还原反应混合物中,封端副产物浓度被维持低于如下浓度:在该浓度下,再封端阻止了工程化半胱氨酸残基被进一步活化。所述方法提供一种非封端的半胱氨酸蛋白质制剂。典型地,采用所述方法,至少60%的半胱氨酸是脱封端(或者,换言之,是活化的)。在某些方面,采用所述方法,至少70%,至少75%或者至少80%的封端的半胱氨酸残基是被脱封端的。在某些方面,采用本发明方法,蛋白质分子中至少85%的还原反应前就存在的链间二硫键,在脱封端工程化的半胱氨酸抗体制剂中仍然保留。在某些方面,选择还原剂和还原条件,使得不超过20%,不超过15%,不超过10%或者不超过5%的蛋白质分子中存在的链间二硫键,在还原反应过程中被转化成一对自由巯基。
附图说明
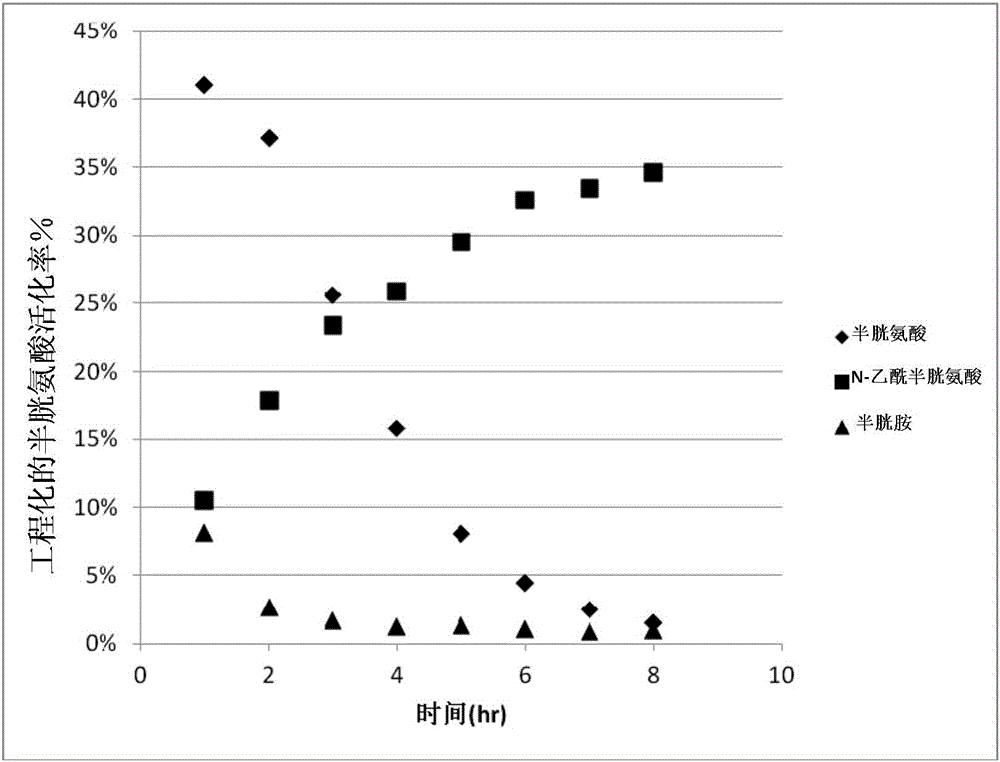
图1显示了用三种不同的单巯基还原剂(半胱氨酸、N-乙酰半胱氨酸(NAC)和半胱胺),在相同的条件下(低浓度2mM)处理一种ecmAb(溶液浓度99μM),对所得的样品进行rPLRP柱分析的结果。Y轴代表可用的总工程化的半胱氨酸的活化百分比。没有一种还原剂能够达到对可用的工程化的半胱氨酸残基的100%激活。没有证据表明,在这些实验中,链间二硫键被还原。
图2显示了一种S239C ecmAb mcMMAF ADC的重链的rPLRP柱分析结果。S239C ecmAb用本发明中的方法选择性地还原,并且偶联到mcMMAF药物接头。还原剂是0.8mM半胱氨酸。工程化的半胱氨酸残基的活化基本上进行到完成。标注为%H0的柱表示没有与药物接头偶联的重链。标注为%H1的柱表示偶联有1个药物接头的重链;而标注为%H2的柱表示偶联有2个药物-接头的重链。
图3显示了S239C ecmAb mcMMAF ADC重链的rPLRP柱分析结果。S239C ecmAb用本发明中的方法选择性地还原,并且偶联到mcMMAF药物-接头上。还原剂是0.55mM半胱氨酸。只有小百分比的工程化的半胱氨酸残基保持封端(%H0),而未封端工程化的半胱氨酸残基的百分比接近100%(%H1),而且非特异性还原(%H2)的量保持在低位。
图4显示了通过S239C ecmAbs选择性实现了工程化的半胱氨酸的活化,该活化与重链和轻链可变区的种类无关。mAb 1是人源化2H12ecMAb,mAb2是人源化的h1F6ecMab(活化:%H0;选择性:%H2)。对于两种mAbs而言,脱封端速率是类似的。工程化半胱氨酸残基的选择性活化,在mAb1的三个变异位点S239,K326和A327上得到证明。这些结果表明,工程化半胱氨酸的活化可以在不同位点选择性地实现。这些实验中采用约0.9mM的半胱氨酸。
图5显示了mAb1的S239C突变体的工程化的半胱氨酸的活化。反应混合物中mAb的浓度与图4反应中类似,但是半胱氨酸的浓度更低一些,在整个时间进程中大约为0.5mM。这个实验显示,采取低浓度的半胱氨酸可获得最大选择性,并且在给予足够时间的情况下,可以以非常高的选择性实现完全活化。这次实验最终时间点所制备得到的偶联物的平均载药量是1.93,这与标称值2.0很接近,并且落在通过传统非选择性还原然后再氧化所得的数值范围内。
发明详细描述
Ⅰ.综述
在本发明中,提供了一种选择性移除抗体(包括单克隆抗体)中工程化的半胱氨酸上硫化物封端的方法。尽管在将抗体长时间暴露于低浓度小分子硫醇中后观察到某些选择性移除,但是在静态反应混合物中不会发生完全的脱封端。有利地,用本发明的方法,其中当还原副产物被移除时维持还原剂浓度,能够实现工程化的半胱氨酸的近乎完全活化。在不需要活化基本上所有工程化半胱氨酸的实施例中,本方法可以用于控制活化水平。令人意外的是,获得的脱封端的抗体具有完整的链间二硫键,从而维持了抗体结构,并消除了在将抗体与药物或者其它功能剂进行偶联前的再氧化步骤。通过本发明方法,对制备抗体偶联物(包括抗体药物偶联物)的目前生产操作进行了大幅简化。
Ⅱ.定义
如本文所用,术语“抗体”泛指完整的单克隆抗体、多克隆抗体、单特异性抗体、多特异性抗体(例如双特异性抗体),和那些表现出所需生物活性(例如特异性结合到目标抗原)和那些至少具有一个天然链间二硫键的抗体片段。典型的片段包括,例如,Fabs,迷你抗体(minibodies)及其类似物。一个完整的抗体通常由4个多肽链组成(两个重链和两个轻链),每个多肽主要有2个区域:可变区和恒定区。可变区特异性地结合于目标抗原并与目标抗原反应。可变区包括互补决定区(CDRs),所述互补决定区识别和结合到某一特定抗原的特异性结合位点。恒定区可以被免疫系统识别和发生相互作用(见例如,Janeway et al.,2001,Immuno.Biology,第5版,Garland Publishing,New York)。这4个多肽通过链间二硫键相互共价连接。抗体可以是任何型(例如IgG,IgE,IgM,IgD和IgA),类别(例如IgG1,IgG2,IgG3,IgG4,IgA1和IgA2)或亚类。抗体可以来自任何合适的物种。在某些实施方式中,抗体是人或者鼠来源的。单克隆抗体可以是例如,人的,人源化的,或者嵌合的。根据上下文,术语“抗体”可以指一个单一的抗体分子,或者抗体分子的集合,例如在抗体溶液中。
如本文所用,术语“链间二硫键”指一个抗体相邻多肽链上的两个半胱氨酸残基之间的共价键。二硫键具有结构式R1-S-S-R2,其中S原子位于半胱氨酸的侧链,R1和R2代表半胱氨酸残基的其余部分以及所在的多肽链。链间二硫键通常位于一个抗体分子的重链和轻链之间,或者两条重链之间。
如本文所用,术语“工程化的半胱氨酸残基”指被引入到蛋白质(例如抗体)多肽序列中的半胱氨酸残基。含有半胱氨酸残基的单克隆抗体,可以称为“ecmAb”。工程化的半胱氨酸残基通常在蛋白质的天然(自然存在的)肽序列中是不存在的。工程化的半胱氨酸残基可以取代多肽序列某一给定位点的自然存在的氨基酸,并通过重组技术例如定点诱变被引入到多肽序列上。工程化的半胱氨酸残基可以是封端或者非封端的。
如本文所用,术语“非封端(或脱封端)半胱氨酸残基”指一半胱氨酸残基,其α侧链含有一个自由的巯基,其具有结构R1-SH。R1代表半胱氨酸残基的非巯基部分。非封端半胱氨酸残基可以是非封端的工程化的半胱氨酸残基。
如本文所用,术语“封端的半胱氨酸残基”指一个半胱氨酸残基,其α侧链含有一个二硫键化部分,其具有结构式R1-S-S-R3。R1代表半胱氨酸残基的非巯基部分,并且R3代表分子量小于或等于500Da的封端剂的非巯基部分。封端剂可以是,例如,半胱氨酸、半胱氨酰甘氨酸、或者谷胱甘肽(R3分别代表自由半胱氨酸、半胱氨酰甘氨酸的非巯基部分,或者谷胱甘肽的非巯基部分)或者其它可用的单硫醇。封端半胱氨酸残基可以是封端的工程化的半胱氨酸残基。
如本文所用,术语“还原反应”指这样的反应,其中具有结构式R1-S-S-R3的封端半胱氨酸残基(例如封端的工程化的半胱氨酸残基)被还原,并且形成具有结构R1-SH的硫醇部分。R1和R3如上定义。
如本文所用,术语“还原剂”指能够还原二硫键的化合物。还原剂包括但不限于:硫醇例如半胱氨酸、半胱胺、和β-硫基乙醇。
如本文所用,术语“氧化剂”指能够将一对自由巯基转化为二硫键的化合物。氧化剂的例子包括但不限于:5,5′-二硫代(2-硝基苯甲酸)(DTNB)、脱氢抗坏血酸(DHAA)和硫酸铜(CuSO4)。“再氧化步骤”是使得一对自由巯基转化为二硫键的确定性步骤。该确定性步骤包括:引入外源氧化剂和/或有意的保持一段时间,以便发生自氧化。
如本文所用,从包含抗体和某一物质的混合物中术语“移除”该物质(例如封端副产物),是指从混合物中移除该物质的任一部分,包括该物质的全部。移除这种物质还可以包括:将所述抗体从包含该物质的第一混合物中转移到不包含该物质的第二混合物中。从混合物中移除某一物质,可包括步骤例如透析、渗滤、色谱法等。
如本文所用,术语“抗体药物偶联物”和“ADC”指可任选地通过接头而偶联有治疗剂(例如药物)的抗体。
如本文所用,术语“药物-接头化合物”,或“药物-接头”指具有药物部分和连接在其上的接头的分子,其中所述接头含有适合于连接到抗体中的氨基酸残基(例如半胱氨酸残基)的反应性基团。
Ⅲ.具体实施方式描述
在本发明中,提供了一种选择性还原工程化的半胱氨酸抗体从而形成非封端抗体制剂的方法。这种方法包括:将还原试剂与工程化的半胱氨酸抗体分子接触,其中每个抗体分子具有至少一个封端的工程化的半胱氨酸残基和至少一个链间二硫键,以及将还原剂与抗体分子在足以使工程化的半胱氨酸残基脱封端并形成封端副产物的条件下进行反应。在还原反应过程中,封端副产物被移除,从而阻止新形成的硫醇被再次发生封端。随着封端副产物一起被移除的还原剂,被替换以驱动还原反应向前进行。选择还原剂和还原条件,使得工程化的半胱氨酸残基被选择性地还原(即,选择性地活化)。因此,不需要为了重新形成链间二硫键而在还原步骤之后采取单独的再氧化步骤。具有工程化的半胱氨酸残基的抗体分子中的基本上所有链间二硫键,在脱封端的抗体中都被保留。使用术语“保留”,并不表示链间二硫键在还原反应中保持不变。键在还原反应过程中可以被破坏,但是在转化为一对自由巯基之前重新形成键。键的重新形成不依赖于单独的再氧化步骤,而是在还原反应过程中发生,如图2所示。
流程1显示了采用本发明的方法,对半胱氨酸残基进行脱封端的示意图,其中用单克隆抗体作为示范的蛋白质。如反应(i)所示,还原剂(1)与具有封端工程化的半胱氨酸残基(2)的抗体反应,结果产生具有脱封端的半胱氨酸残基的抗体(3)和封端副产物(4)。反应(i)能够向两个方向进行,并且脱封端的工程化氨基酸残基与封端副产物之间反应,能够导致工程化的半胱氨酸残基被再次封端。逆反应的结果是形成一组合物,在该组合物中只有一部分抗体具有脱封端的工程化的半胱氨酸残基。根据本发明方法,向反应混合物中补充额外的还原剂,如在反应(ii)所示,并且将反应副产物从反应混合物中移除。补充还原剂和移除反应副产物能够驱动正反应和防止逆反应,从而促使形成所需的具有脱封端工程化的半胱氨酸残基的抗体(3)。
流程1
在正常(即“静态溶液”)条件下,在再封端反应限制了工程化的半胱氨酸被活化的程度。图1中的图显示了在相同条件下用三种不同的单硫醇处理ecmAb的结果。Y轴代表总的可用的工程化的半胱氨酸活化的百分比。这张图显示:每种还原剂都不能活化近乎100%的可用的工程化的半胱氨酸,而这种限制是因为二硫键副产物(流程1中的4)会将生成的硫醇再封端,如上所述。
还原剂
任何合适的还原剂都可以用于本发明的方法。还原剂的例子包括但不限于:单硫醇还原剂例如半胱氨酸、N-乙酰半胱氨酸、半胱胺、β-巯基乙醇、2-巯基乙磺酸钠盐等。温和的还原剂例如半胱氨酸等特别适合于移除封端半胱氨酸残基(例如封端的工程化的半胱氨酸残基)的封端,并不破坏链间二硫键。强还原剂,例如二硫苏糖醇(DTT)、二硫赤藓糖醇(DTE)和双(2-巯基乙基)砜,在大多数情况下,能够过快还原链间二硫键。具体地,对于还原剂例如TCEP和DTT是不可能选择性地使封端半胱氨酸的发生脱封端,对于所述还原剂而言流程2中混合二硫键化物的中间产物6或者不形成,或者仅仅暂时形成。在一些实施方式中,还原剂选自半胱氨酸、半胱胺、β-巯基乙醇、2-巯基乙磺酸钠盐及其混合物。
反应条件
尽管多种单硫醇可以用于选择性活化,图1中的图表显示,不同的硫醇以不同的速率活化工程化的半胱氨酸,并达到不同的最大化程度。因此,确定工程化的半胱氨酸残基活化的合适条件,包括确定合适的还原剂和确定合适的还原剂浓度。
反应条件的确定,部分地是根据工程化的半胱氨酸的位置,封端剂的种类,或者特定抗体上的残基。例如,溶剂暴露的工程化的半胱氨酸残基,较之被掩埋或者部分掩埋的工程化的半胱氨酸残基,更容易被脱封端。
有利于选择性还原蛋白中非配对半胱氨酸残基的条件,可以基于流程2中的反应来理解。如果还原剂与工程化的半胱氨酸残基反应(如在反应(a)所示)比其与链间二硫键(如在反应(b)所示)的反应快得多,那么选择性活化是相对简单的。这种情形仅仅只发生在,如果有的话,半胱氨酸高度暴露于抗体表面的溶剂。然而,经常地,在抗体偶联物例如ADCs中使用的ecmAbs中的工程化的半胱氨酸而言,低暴露的位点是优选的。还原剂可以与这些低暴露的工程化的半胱氨酸残基以及与二硫键以可比较的速率进行反应。因此,当反应(a)和反应(b)以可比较的速率发生时,常常需要找到选择性的活化条件。
在反应(a)和反应(b)以可比的速率发生—或者反应(b)比反应(a)更快的情形下,选择性活化工程化的半胱氨酸残基就要求确保当链间二硫键被还原剂攻击的时候,重新形成二硫键的反应(c)要快于第二还原步骤(d),该步骤(d)会导致不希望的链间二硫键的断裂。通常,不可能影响反应(c)的速率,因为它是单分子反应。硫醇和混合二硫键产物(如6所示)是同一抗体的部分。但是反应(d)的速率取决于还原剂的浓度,因此可以通过使用一个足够低浓度的还原剂使得反应(d)比反应(c)慢。当然,反应(a)和反应(b)也取决于还原剂的浓度,所以在这种条件下,所述活化也很慢。因此,通过在一段长的时间内维持非常温和的还原条件(低还原剂浓度和相对温和的还原剂),通常有利于选择性活化工程化的半胱氨酸。
流程2
还原反应混合物可以包括任何合适量的蛋白质。典型地,在还原反应混合物中蛋白质浓度(不管是抗体还是非抗体蛋白质)的浓度范围为:大约0.01mg/mL到大约150mg/mL,更典型地,从大约1mg/ml到大约50mg/ml。所述还原反应混合物可以包含,例如:大约0.01、0.05、0.1、0.25、0.5、1.0、2.0、3.0、4.0、5.0、6.0、7.0、8.0、9.0、10.0、12.5、15、17.5、20、22.5、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100、110、120、130、140或大约150mg蛋白质(不管是抗体还是非抗体蛋白质)/每mL还原反应混合物。蛋白质浓度(不管是抗体还是非抗体蛋白质)可以更高或更低,这取决于采用的特定反应条件。一个本领域技术人员能够将质量浓度(例如mg/mL)换算为摩尔浓度(例如摩尔/L)。
任何合适量的还原剂可以应用于本发明的方法。通常,还原反应混合物中还原剂浓度足够高以至于还原反应能够发生,但是足够低以至于链间二硫键的还原缓慢到几乎可以忽略。典型地,采用一起始量的还原剂在起始浓度下,从而形成还原反应混合物。通过在还原反应持续过程中连续或分批向还原反应混合物补充额外的还原剂,基本上维持这种起始浓度。在某一实施方式中,在整个还原反应过程中,以连续方式向还原反应混合物中补充额外的还原剂。优化还原剂浓度可以采用这里描述的教导通过实验加以确定,并且对不同的硫醇可以是不同的,因为它们的还原能力不同。已确定,在实施例所描述的条件下,半胱氨酸的最优浓度为大约0.5mM到大约1.5mM。依据还原剂的还原能力和ecmAb的浓度,这一浓度可以提高或降低。在某些实施例中,还原剂的浓度被维持在高于总抗体的浓度。最优的还原剂相对于总抗体的浓度比,也取决于还原剂的还原能力。在某些方面,还原反应混合物中还原剂的浓度,比还原反应混合物中总抗体浓度高大约5倍到大约25倍,5倍到大约20倍,5倍到大约15倍,5倍到大约10倍。例如,在某些实施方式中,还原反应混合物中还原剂与总抗体的浓度比为大约5∶1、6∶1、7∶1、8:1、9∶1、10∶1、11∶1、12∶1、13∶1、14∶1、15∶1、16∶1、17∶1、18∶1、19∶1,或20∶1。在某些实施方式中,包括某些实施方式中,当还原剂是半胱氨酸的时候,还原反应混合物中还原剂与总抗体浓度比为从5∶1到大约12∶1,从7∶1到大约12∶1,优选地大约8∶1。在一些方面,工程化的半胱氨酸残基位于重链的239位(命名根据欧洲索引)。其它浓度比也可应用于本发明的方法,这取决于多种因素,例如采用的特定的还原剂,或者工程化的半胱氨酸在抗体上的位置。
进行半胱氨酸残基(工程化的或者天然的半胱氨酸残基)脱封端的还原反应,从而减少链间二硫键的还原和后续自由巯基的产生。采用本发明方法形成的脱封端蛋白质制剂中,在具有封端半胱氨酸残基的蛋白质上存在的基本上所有链间二硫键都被保留。通常,至少约80%链间二硫键被保留在脱封端蛋白质制剂中。在某些实施方式中,至少大约85%链间二硫键被保留在脱封端蛋白质制剂中。在某些实施方式中,至少大约90或者大约95%链间二硫键被保留在脱封端蛋白质制剂中。在某些实施方式中,所有链间二硫键被保留在脱封端蛋白质制剂中。或者,换言之,在代表性的实施方式中,不超过大约20%,不超过大约15%,不超过大约10%,或者不超过大约5%的链间二硫键在还原反应中被转化为一对自由巯基。当它应用于抗体时,存在于具有封端工程化的半胱氨酸残基的抗体分子(例如还原反应前的抗体分子)上的基本上所有链间二硫键,都被保留在用本发明方法形成的脱封端抗体制剂中。通常,脱封端抗体制剂中,至少约80%的链间二硫键被保留。在某些实施方式中,脱封端抗体制剂中,至少约85%的链间二硫键被保留。在某些实施方式中,脱封端抗体制剂中,至少约90%或者大约95%的链间二硫键被保留。在某些实施方式中,脱封端抗体制剂中,全部的链间二硫键被保留。或者,换言之,在代表性实施方式中,不超过大约20%,不超过大约15%,不超过大约10%,或者不超过大约5%的链间二硫键,在还原反应中被转化为一对自由巯基。
在还原反应混合物中还原剂浓度被维持时,还原副产物从还原反应混合物中被移除。还原剂的添加和副产物的移除可以连续或分步的方式进行。在某些实施方式中,还原剂的添加和副产物的移除以连续的方式进行。还原剂的添加和副产物的移除可以采用技术包括但不限于:切向流过滤(例如渗滤)、尺寸排阻色谱法、固相固定法和透析。在渗滤过程中,还原剂水溶液通常是以给定的速率添加到还原反应混合物中,而一部分混合物以同样的速率被移除。半透膜可以用于将蛋白质(例如抗体)保留在还原反应混合物中。或者,所述蛋白质(例如抗体)可以被固定在固相载体上(例如蛋白质A的琼脂糖珠),而水相还原剂溶液可以流经固定化的蛋白质(例如抗体)。也可以使用能够和封端副产物反应的活性树脂。例如,封端副产物可以用具有反应性基团(例如孔内具有二硫键产物)的树脂进行捕获,其中这些孔足够小从而将蛋白质(例如抗体)排除在外。
相应地,本发明一些实施方式中提供了如上所述的方法,其中,从还原反应混合物中移除封端副产物包括:在还原反应过程中,透析或者渗滤所述的还原反应混合物。在一些实施方式中,从还原反应混合物中移除封端副产物包括:在还原反应过程中,渗滤所述的还原反应混合物。
通常,实施本发明方法为了最大化进行半胱氨酸残基的脱封端。然而,任一种蛋白质分子可具有封端的半胱氨酸残基以及非封端的残基。通常,脱封端抗体制剂将包含两种或两种以下物质:一种具有至少二个脱封端工程化的半胱氨酸残基且没有封端工程化的半胱氨酸残基的抗体分子;一种具有至少二个封端工程化的半胱氨酸残基且没有脱封端工程化的半胱氨酸残基的抗体分子;和一种具有至少一个封端工程化的半胱氨酸残基和至少一个脱封端工程化的半胱氨酸残基的抗体分子。有利的是,本发明方法提供高水平的选择性脱封端工程化的半胱氨酸残基。例如,执行所述方法以至于至少40%的工程化的半胱氨酸残基被脱封端。在某些方面,执行所述方法以至于至少40%,至少大约45%,至少大约50%,至少大约55%,至少大约60%,至少大约65%,至少大约70%,至少大约75%,至少大约80%,至少大约85%,至少大约90%的工程化的半胱氨酸残基被脱封端。在某些实施方式中,至少60%的工程化的半胱氨酸残基被脱封端。高或者低水平的脱封端都可能发生,这部分取决于特定抗体以及工程化的半胱氨酸残基在抗体上的位置。尽管,本发明的方法通常被执行,从而使得半胱氨酸残基的脱封端最大化,但是在有些情况下,使半胱氨酸残基脱封端最大化并不需要,例如,当一个抗体具有二个半胱氨酸残基,并只需要将一个半胱氨酸残基与功能性试剂偶联的时候。在某些这样的实施方式中,本发明方法可以用于严格控制工程化的半胱氨酸的活化程度。本发明方法的有利的方面是,中等程度的脱封端很容易实现,只要将活化停止在所需要的水平并进行偶联即可。当采用全还原后再氧化的方法时,难以获得这种中等水平的偶联物。
反应混合物可以包含额外的反应试剂或化合物。作为非限制性例子,反应混合物可以包含缓冲剂(例如2-(N-吗啉)乙磺酸(MES),2-[4-(2-羟乙基)-1-哌嗪基]乙磺酸(HEPES),3-吗啉丙磺酸(MOPS),三羟甲基氨基甲烷(TRIS),磷酸钾,磷酸钠,磷酸盐缓冲液,柠檬酸钠,乙酸钠,和硼酸钠),共溶剂(例如二甲亚砜、二甲基甲酰胺、乙醇、甲醇、异丙醇、丙三醇、四氢呋喃、丙酮、乙腈和乙酸),盐(例如NaCl,KCl,CaCl2,Mn2+和Mg2+的盐),变性剂(例如尿素和盐酸胍),清洁剂(例如十二烷基磺酸钠,和Triton-X100),和螯合剂(例如乙二醇双(2-氨基乙醚)-N,N,N’,N’-四乙酸(EGTA)、2-({2-[双(羧甲基)氨基]乙基}(羧甲基)氨基)乙酸(EDTA),以及1,2-双(邻氨基苯氧基)乙烷-N,N,N’,N’-四乙酸(BAPTA))。缓冲溶液、共溶剂、盐、变性剂、清洁剂和螯合剂可以采用任何合适的浓度,这些对于本领域技术人员而言是很容易确定的。通常,如果存在缓冲剂、共溶剂、盐、变性剂、清洁剂和螯合剂,其被包含在在反应混合物中的浓度范围为大约1μM到大约1M。例如,缓冲剂、共溶剂、盐、变性剂、清洁剂和螯合剂可以被包含在反应混合物中,其浓度为大约1μM,或大约10μM,或大约100μM,或大约1mM,或大约10mM,或大约25mM,或大约50mM,或大约100mM,或大约250mM,或大约500mM,或大约1M。具体地,共溶剂可以被包含在反应混合物中,其含量范围为,例如大约1%v/v到大约75%v/v,或更高。共溶剂可以被包含在反应混合物中,例如含量为大约5,10,20,30,40或50%v/v。
在足以形成脱封端半胱氨酸残基的条件下进行反应。反应可以在任何合适温度下进行。通常,反应在大约4℃到大约40℃的温度范围内进行。反应可以在例如,大约25℃到大约37℃的温度范围内进行。反应可以在任何pH条件下进行。通常,反应在大约6.5到大约10的pH范围内进行。在某些情况下,pH范围为大约7.0到大约8.5。反应可以在任何合适的时长下进行。时长的选择取决于还原剂的能力。因为温和的还原条件(低浓度还原剂和相对温和的还原剂)被应用于本发明方法,因此还原反应将会延长到大于典型的还原链间二硫键(所需)预期时间。在一些优选的方面,还原反应将会继续直到至少大约60%,至少大约70%或至少大约80%或至少大约85%封端的工程化的半胱氨酸残基被脱封端。在某些方面,还原反应在合适的条件下温育至少1h,至少2h,至少3h,至少4h,至少5h,或者至少6h。反应可以在惰性气体氛围下进行,例如氮气氛围或者氩气氛围。其它的反应条件可应用于本发明,这取决于某一特定抗体的种类,或者还原剂。
在某些实施方式中,反应混合物的pH范围从大约6.5到大约8.5。在某些实施方式中,本发明方法包括:维持反应混合物的温度在大约4℃到大约37℃的范围。在某些实施方式中,反应混合物在期望的温度下维持一段时间,从大约1h到大约8h。
一些已知的纯化技术可以应用于本发明方法中的不同节点。这些技术可以根据需要,被用于移除过量的还原剂,将缓冲溶液或者其它组分更换入或者更换出所述的反应混合物,以及浓缩或者稀释抗体组分。用于本发明方法的纯化技术包括但不限于:切向流过滤(TFF),凝胶过滤,免疫沉淀,亲和色谱法等。优选地,减少纯化时间,从而防止不必要的氧化或者脱封端工程化的半胱氨酸残基被再封端。
抗体偶联物
抗体上脱封端工程化的半胱氨酸残基可充当有用的基座,以用于安装多种功能基团,其中包括成像试剂(例如发色基团和荧光基团),诊断试剂(例如MRI对比剂和放射性同位素),稳定剂(例如乙二醇聚合物)和治疗剂。具有脱封端半胱氨酸残基的抗体可以被偶联到功能剂以形成抗体-功能剂的偶联物。功能剂(例如药物,检测试剂,稳定剂)被偶联(共价连接)至抗体上的工程化的半胱氨酸残基位点。功能剂可以通过功能剂上的活性巯基直接地、或者是通过接头间接地进行连接。
具有脱封端的半胱氨酸残基的抗体,可以偶联药物从而形成抗体药物偶联物(ADCs)。典型地,ADC包含位于药物和抗体之间的接头。接头可以是可降解的或者是不可降解的接头。可降解的接头典型地在细胞内环境下容易降解,例如在目标位点处接头发生降解,从而使药物从抗体上释放出来。合适的可降解的接头包括,例如酶降解的接头,其中包括可以被细胞内蛋白酶(例如溶酶体蛋白酶或者内体蛋白酶)降解的含有肽基的接头,或者糖接头例如,可以被葡糖苷酸酶降解的含葡糖苷酸的接头。肽基接头可以包括,例如二肽,例如缬氨酸-瓜氨酸,苯丙氨酸-赖氨酸或者缬氨酸-丙氨酸。其它合适的可降解的接头包括,例如,pH敏感接头(例如pH小于5.5时水解的接头,例如腙接头)和在还原条件下会降解的接头(例如二硫键接头)。不可降解的接头典型地在抗体被蛋白酶水解的条件下释放药物。
连接到抗体之前,接头具有能够和脱封端的工程化的半胱氨酸残基反应的活性反应基团,连接通过活性反应基团实现。巯基特异性的活性反应基团是优选的,并包括:例如马来酰亚胺类化合物,卤代酰胺(例如碘、溴或氯代的);卤代酯(例如碘、溴或氯代的);卤代甲基酮(例如碘、溴或氯代),苄基卤代物(例如碘、溴或氯代的);乙烯基砜,吡啶基二硫化物;二硫化二氧化衍生物(disulfide dioxide derivatives);汞衍生物例如3,6-二-(汞甲基)二氧六环,而对离子是醋酸根、氯离子或者硝酸根;和聚亚甲基二甲基硫醚硫代磺酸盐。接头可以包括,例如,通过硫代丁二酰亚胺连接到抗体上的马来酰亚胺。
药物可以是任何细胞毒性,抑制细胞生长或者免疫抑制的药物。在实施方式中,接头连接抗体和药物,而药物具有可以和接头成键的功能性基团。例如,药物可以具有可以和连接物成键的氨基,羧基,巯基,羟基,或者酮基。在药物直接连接到接头的情况下,药物在连接到抗体之前,具有能够与脱封端的半胱氨酸残基反应的活性基团。
有用的药物类别包括,例如,抗微管蛋白药物、DNA小沟结合试剂、DNA复制抑制剂、烷化试剂、抗生素、叶酸拮抗物、抗代谢药物、化疗增敏剂、拓扑异构酶抑制剂、长春花生物碱等。特别有用的细胞毒性药物类的例子包括,例如,DNA小沟结合试剂、DNA烷基化试剂、和微管蛋白抑制剂、典型的细胞毒性药物包括、例如奥瑞他汀(auristatins)、喜树碱(camptothecins)、多卡霉素/倍癌霉素(duocarmycins)、依托泊甙(etoposides)、美登木素(maytansines)和美登素类化合物(maytansinoids)(例如DM1和DM4)、紫杉烷(taxanes)、苯二氮卓类(benzodiazepines)或者含有苯二氮卓的药物(benzodiazepine containing drugs)(例如吡咯并[1,4]苯二氮卓类(PBDs),吲哚啉苯并二氮卓类(indolinobenzodiazepines)和噁唑烷并苯并二氮卓类(oxazolidinobenzodiazepines))和长春花生物碱(vinca alkaloids)。选择的含有苯并二氮卓类的药物在WO 2010/091150,WO 2012/112708,WO 2007/085930,和WO 2011/023883中有描述。
在某些典型的实施方式中,合适的细胞毒性试剂包括:DNA小沟结合试剂(例如,烯二炔类(enediynes)和莱希菌素(lexitropsins)、CBI化合物;见美国6,130,237号专利),多卡霉素/倍癌霉素(duocarmycins)(见美国专利公开号为20060024317)、紫杉烷(例如紫杉醇(paclitaxel)和多烯紫杉醇(docetaxel))、嘌呤霉素(puromycins)、长春花生物碱、CC-1065,SN-38、拓扑替康(topotecan)、吗啉代-阿霉素/多柔比星(morpholino-doxorubicin)、根霉素(rhizoxin)、氰基吗啉基代-阿霉素/多柔比星(cyanomorpholino-doxorubicin)、棘霉素(echinomycin)、考布他汀(combretastatin)、纺锤菌素(netropsin)、埃博霉素(epothilone)A和B、雌莫司汀(estramustine)、念珠藻素(cryptophysins)、西马多丁(cemadotin)、美登素类化合物(maytansinoids)、迪斯德莫来/圆皮海绵内酯(discodermolide)、艾榴塞洛素(eleutherobin)和米托蒽醌(mitoxantrone)。
所述药物可以是抗微管蛋白药物(anti-tubulin agent)。抗微管蛋白药物的例子包括,但不限于,紫杉烷(例如(紫杉醇),泰索帝/多西(多烯紫杉醇)),T67(杜拉瑞克(Tularik))和长春花生物碱(例如长春新碱(vincristine),长春花碱(vinblastine),长春地辛(vindesine)和长春瑞滨(vinorelbine))。其它抗微管蛋白药物包括,例如浆果赤霉素衍生物(baccatinderivatives)、紫杉烷类似物(例如埃博霉素A和B)、诺考达唑(nocodazole)、秋水仙碱(colchicine)和秋水仙胺(colcimid)、雌莫司汀(estramustine)、念珠藻素(cryptophysins)、西马多丁(cemadotin)、美登素类化合物(maytansinoids)、考布他汀(combretastatins)、迪斯德莫来/圆皮海绵内酯(discodermolide)、奥瑞他汀(auristatins)、和艾榴塞洛素(eleutherobin)。
药物可以是美登木素或美登醇,另外类别的抗微管蛋白药物(ImmunoGen,Inc.;还可参见Chari等人,1992,Cancer Res.52∶127-131以及U.S.专利No.8,163,888)。
药物可以是奥瑞他汀(auristatin)。奥瑞他汀(Auristatins)包括,但不限于,AE,AFP,AEB,AEVB,MMAF,和MMAE。奥瑞他汀(Auristatins)的合成和结构描述见美国专利申请公开号2003-0083263和2009-0111756;国际专利申请公开号WO 04/010957;国际专利申请公开号WO 02/088172;美国专利号6,884,869;美国专利号7,659,241;美国专利号7,498,298;美国专利号8,343,928;和美国专利号8,609,105;出于各种目的,每一篇通过引用方式整体引入。
在某些实施方式中,药物部分选自下组:抗微管蛋白试剂,DNA结合试剂,和DNA烷化试剂。在某些实施方式中,药物选自下组:奥瑞他汀(auristatin),吡咯并苯并二氮卓类(pyrrolobenzodiazepine),多卡霉素/倍癌霉素(duocarmycin),美登醇,紫杉烷,卡奇霉素(calicheamicin)和蒽环霉素(anthracycline)。
药物-接头可以用于在一个简单步骤中形成ADC。在其它实施方式中,双功能连接物化合物可以用于在两步或多步方法中形成ADC。例如,脱封端的工程化的半胱氨酸残基在第一步骤中与接头的反应活性部分反应,并且在随后的步骤中,接头上的功能性基团与药物反应,从而形成ADC。
通常,选择接头上功能性基团,以利于特异性地与药物部分上的合适的反应活性基团进行反应。作为非限制性的例子,基于叠氮化合物的部分可以用于特异性地与药物部分上的反应性炔基基团反应。药物通过叠氮和炔基之间的1,3-偶极环加成,从而共价结合于接头。其它的有用的功能性基团包括,例如酮类和醛类(适合与酰肼类和烷氧基胺反应),膦(适合与叠氮反应);异氰酸酯和异硫氰酸酯(适合与胺类和醇类反应);和活化的酯类,例如N-羟基琥珀酰亚胺酯(适合与胺类和醇类反应)。这些和其它的连接策略,例如在《生物偶联技术》,第二版(Elsevier)中所描述的,是本领域技术人员所熟知的。本领域技术人员能够理解,对于药物部分和接头的选择性反应,当选择了一个互补对的反应活性功能基团时,该互补对的每一个成员既可以用于接头,也可以用于药物。
相应地,本发明的某些实施例提供了制备如上所述的脱封端抗体制剂的方法,其可进一步地包括:将脱封端抗体(例如将脱封端抗体制剂)与药物-接头化合物,在足以形成抗体偶联物(ADC)的条件下进行结合。
在某些实施方式中,本发明方法包括:在足以形成抗体-接头偶联物的条件下,将脱封端抗体与双功能接头化合物进行结合。在这些实施方式中,本发明方法还进一步地包括:在足以将药物部分通过接头共价连接到抗体的条件下,将抗体接头偶联物与药物部分进行结合。
在某些实施方式中,抗体药物偶联物ADC如下分子式所示:
其中:
Ab是抗体,
LU是接头;
D是药物;
而且下标p是选自1到8的值。
在上式中,接头LU,通过脱封端的工程化的半胱氨酸残基连接到抗体上。下标p的数值取决于可用于偶联的脱封端半胱氨酸残基的数量。例如,对于一个具有2个脱封端的工程化半胱氨酸残基的抗体(例如每个重链上有1个或者每个轻链上有1个位点),p值可以是2。类似地,对于一个具有4个脱封端的工程化半胱氨酸残基的抗体(例如每个重链上有2个位点,或者每个轻链上有2个位点),p值可以是4。在某些优选的实施方式中,p值为1到4的值。
载药量
抗体上药物-接头分子的平均数量(或者平均载药量)是ADC组分的一个重要特性,因为它基本决定了能够输送到目标细胞的药物量。平均载药量包括偶联到工程化的半胱氨酸残基的药物,还有偶联到目标工程化的半胱氨酸残基之外的其它位点的药物,以及在组合物中未偶联的抗体的量。如果目标是平均载药量为每个抗体有2个药物,那么可以用具有2个半胱氨酸残基的抗体(例如每个重链上1个位点或者每个轻链上1个位点)来制备ADC组分。如果目标是平均载药量为每个抗体有4个药物,那么可以用具有4个半胱氨酸残基的抗体(例如每个重链上2个位点或者每个轻链上2个位点,或者每个重链上1个位点并且每个轻链上1个位点)来制备ADC组分。本领域技术人员能够理解,治疗用的其它载药量水平取决于特定抗体或特定药物(包括,例如,载药量小于2或者载药量大于4)。用于药物偶联的结合位点可以被引入抗体,这可以通过在重链上1个以上或者2个以上位点设置工程化的半胱氨酸残基,或者在轻链上设置工程化的半胱氨酸残基,或者兼而有之。重要的是,上述工程化的半胱氨酸残基脱封端水平,导致可以制备具有有用载药量的ADC组合物。
典型地,采用具有2个工程化的半胱氨酸残基的抗体所制备的ADC组合物,其平均载药量为每个抗体有大约1.5到2.5个药物。每个抗体上的平均药物量可以是,例如,大约1.5,1.6,1.7,1.8,1.9,2.0,2.1,2.2,2.3,2.4,或2.5。在某些实施方式中,采用具有2个工程化的半胱氨酸残基的抗体所制备的ADC组合物,其平均载药量为每个抗体有大约1.5到大约2.2个药物。典型地,采用具有4个工程化的半胱氨酸残基的抗体所制备的ADC组合物,其平均载药量为每个抗体有大约3.4到大约4.5个药物。每个抗体上的平均药物数量可以是,例如,大约3.3,3.4,3.5,3.6,3.7,3.8,3.9或4.0。在某些实施方式中,采用具有4个工程化的半胱氨酸残基的抗体所制备的ADC组合物,其平均载药量为每个抗体有大约3.6到大约4.2个药物,或者每个抗体有从大约3.8到大约4个药物。
典型地,实施本发明方法,以便选择性修饰未封端的工程化的半胱氨酸残基,但是常常观察到不同程度的非工程化的(例如天然)半胱氨酸残基的修饰。根据需要,可以控制反应条件以限制非工程化的半胱氨酸残基的修饰。在某些特定情形下,可以实行所述方法以消除或者减低非工程化的半胱氨酸残基的修饰。例如,实行所述方法以至于ADC组合物中不超过大约20%,不超过大约15%,不超过大约10%,不超过大约5%的抗体分子中,药物部分被共价连接到非工程化的半胱氨酸残基上。也可以采用上述方法制备ADC组合物,其中具有修饰的非工程化的半胱氨酸残基的抗体数量不超过抗体分子总量的大约5%,10%,15%,20%,25%,或30%。
可以采用多种分析方法确定偶联物的产量和同分异构体混合物。在将药物偶联到抗体之后,偶联的药物-抗体种类可以被分离出来。在某些实施方式中,偶联的抗体种类可以基于抗体、药物和/或偶联物的特性加以分离。其它用于分析ADC组合物的技术包括但不限于:反相色谱,毛细管电泳和质谱。ADC组合物可以例如通过酶解联合LC/MS进行分析,以确定药物部分在ADC上的位置。
抗体
大量的合适的抗体可以用于本发明方法。用于本发明方法的抗体在许多应用领域都有用处,包括体外或者体内诊断,体内成像,以及治疗与独特抗原相关的疾病和症状。5种人的抗体类型(IgG,IgA,IgM,IgD和IgE),和这些类型的多种亚型(例如IgG1,IgG2,IgG3,IgG4,IgA1和IgA2),可基于结构差异进行识别,例如单个抗体分子中免疫球蛋白单元的数量,单个单元上的二硫桥的结构,以及链长度和序列的差异。抗体的类和亚类被称为抗体的同种异型。
抗体可以是完整的抗体或者是抗原结合抗体片段,只要抗体片段含有至少一个链间二硫键。
典型地,抗体是人,啮齿动物(例如小鼠和大鼠),驴,绵羊,兔,山羊,豚鼠,骆驼,马和鸡。抗体可以是,例如,用本领域技术人员熟知技术所制备的鼠的、嵌合的、人源化的或者全人的抗体。重组抗体,例如嵌合的和人源化的单克隆抗体,包括人的和非人的部分,可以通过标准的DNA重组技术获得,它们都是有用的抗体。嵌合抗体是一个分子,其中不同的部分来自不同的动物种,例如具有来自鼠的单克隆抗体的可变区,和来自人免疫球蛋白的恒定区的嵌合抗体(见例如美国专利4,816,567和美国专利4,816,397,在此通过引用方式整体引入本文)。人源化的抗体是指来源于非人物种的抗体分子,具有一个或多个来源于非人物种的互补决定区(CDRs)和来源于人免疫球蛋白分子的框架区域(见美国专利5,585,089,在此通过引用方式整体引入本文)。这些嵌合和人源化的单克隆抗体可以采用本领域熟知的DNA重组技术制备。如本文所用,“人”抗体包括具有人免疫球蛋白序列,以及包括分离自人免疫球蛋白库,分离自人B细胞,或者分离自具有一个或多个人免疫球蛋白的转基因动物(例如美国专利5,939,598和6,111,166中所述)的抗体。
抗体可以是单特异性、双特异性、三特异性、或者更多的多重特异性。
在某些情况下,恒定区具有效应子功能。如本文所用,术语“抗体效应子功能”指一种由Ig的Fc区域贡献的功能。这一功能可以,例如通过Fc效应子区域结合到具有吞噬或裂解活性的免疫细胞上的Fc受体,或者通过Fc效应子区域结合到补体系统中的成分来实现。效应子功能可以是,例如,“抗体依赖的细胞毒性”即ADCC,“抗体依赖的吞噬作用”即ADCP,或者“补体依赖性细胞毒性”即CDC。在某些情况下,恒定区缺乏一个或多个效应功能。通过将药物-接头化合物偶联到效应子功能结合区域内的工程化的半胱氨酸残基,能够调控效应子功能。
抗体可以直接针对任何感兴趣的抗原,例如医学和/或治疗兴趣。例如,抗原可以是与以下有关的抗原:病原体(例如但不限于病毒,细菌,真菌和原生动物),寄生虫,肿瘤细胞,或者特定医疗症状。在肿瘤相关抗原(TAA)情况下,癌症可能是免疫系统的、肺、结肠、直肠、乳腺、卵巢、前列腺、头部、颈部、骨头、或任何其他解剖位置。感兴趣的抗原包括,但不限于,CD30、CD40、LewisY、CD70、CD2、CD20、CD22、CD33、CD38、CD40、CD52、HER2、EGFR、VEGF、CEA、HLA-DR、HLA-Dr10、CA125、CA15-3、CA19-9、L6、Lewis X、甲胎蛋白、CA242、胎盘碱性磷酸酶、前列腺特异抗原、前列腺酸性磷酸酶、表皮生长因子、MAGE-1、MAGE-2、MAGE-3、MAGE-4、转铁蛋白受体抗体、p97、MUC1-KLH、gp100、MART1、IL-2受体、人体绒毛膜促性腺激素、粘蛋白、P21、MPG、和Neu致癌基因产物。
一些具体有用的抗体包括但不限于:针对CD33抗原的抗体(例如,国际专利申请WO 2013/173496所描述的人源化2H12抗体),针对CD70抗原的抗体(例如,国际专利申请WO2006/113909所描述的人源化1F6抗体),针对CD30抗原的抗体(例如,国际专利申请WO2008/025020所描述的人源化AC10抗体),针对CD19抗原的抗体(例如,国际专利申请WO 2009/052431所描述的人源化BU12抗体),针对LIV-1,NTBA,或αVβ6的抗体。许多其它可以和肿瘤特异性抗原结合的内化抗体都可以采用,见综述(参见,例如,Franke等人(2000),Cancer Biother Radiopharm.15∶459-76;Murray(2000),Semin Oncol.27∶64-70;Breitling等人,重组抗体(Recombinant Antibodies),John Wiley,and Sons,New York,1998)。这些参考文献和国际申请中的披露内容在此通过引用引入并用于所有目的。
在某些实施方式中,本发明提供了用于制备具有如上所述具有脱封端的半胱氨酸残基的抗体的方法,其中在抗体中包含至少3个链间二硫键。在某些实施方式中,所述抗体包含至少4个链间二硫键。在某些实施方式中,所述抗体包含1、2、3、4或者5个链间二硫键。在某些实施方式中,工程化的半胱氨酸残基位于抗体的重链恒定区或者轻链恒定区。
工程化的半胱氨酸位置
工程化的半胱氨酸位置可对ADC的性质有影响。例如,全部包埋入蛋白结构的工程化的半胱氨酸残基难以被偶联,因为难以接触溶剂,而位于抗体外表面的工程化的半胱氨酸残基则因为延长暴露于血浆中的物质,导致ADCs具有受损的稳定性。此外,从具有高的表面暴露的工程化的半胱氨酸残基的ecmAbs制备的ADCs,可能对药物的疏水性敏感;而位于更加受保护的位置的工程化的半胱氨酸残基对药物特性更不敏感,因为与溶液中其它物质接触是受限制的。工程化的半胱氨酸残基的位置也可以用于调节效应子功能,这对于一些特定ADC是必要的。例如,将药物-接头偶联到位于效应子功能结合区域的工程化半胱氨酸残基,可以用于封闭与效应子功能调节受体的结合。
在某些实施方式中,工程化的半胱氨酸残基位于重链恒定区、重链可变区、轻链可变区、轻链恒定区、或其组合。优选的半胱氨酸残基是位于这样的位点,所述位点是可偶联的并且导致稳定的连接。“可偶联”的意思是工程化的半胱氨酸残基能够偶联到功能试剂(例如成像试剂,诊断试剂,稳定剂或治疗剂)而不会先使抗体变性。选择引入半胱氨酸残基位点的方法为本领域技术人员熟知,而所述残基随后被偶联于功能性试剂(例如,见,举例来说,Junutula等人,2008,Nature Biotechnology,26(8),925-932)。
在某些方面,工程化的半胱氨酸残基的分数溶剂可及性(fractional solvent accessibility)为10%或者更高,20%或者更高,30%或者更高,40%或者更高,50%或者更高。在某些方面,半胱氨酸残基具有分数溶剂可及性范围从大约10%到95%,从大约10%到85%,从大约10%到75%,从大约10%到60%,从大约20%到95%,从大约20%到85%,从大约20%到75%,从大约20%到60%,从大约40%到95%,从大约40%到85%,从大约40%到75%,从大约40%到60%。确定某一特定位点残基的分数溶剂可及性的方法为本领域熟知并且能够例如采用在线服务器getarea加以确定,该服务器采用Fraczkiewicz和Braun,1998,J.Comp.Chem.,19,319-333(参见http://curie.utmb.edu/getarea.html)中所描述的方法。示例的残基包括位于轻链上的位点15,114,121,127,168,205(根据Kabat编号),或者位于重链的位点112,114,或116(根据Kabat编号法进行编号)。示例的残基包括那些位于IgG1抗体的Fc区域的位点,例如那些位于Fc区域的位点239,326,327或者269(根据EU索引进行编号)。位于位点239,326,327的残基的分数溶剂可及性分别为大约50%,大约94%,和大约23%。
本领域技术人员能够识别,选择性活化工程化的半胱氨酸残基所需要的条件将取决于工程化的半胱氨酸残基在抗体上的位置。流程2的化学式为选择能够选择性活化工程化的半胱氨酸残基的还原条件提供了一个框架,不管它们发生在蛋白序列的什么位置。在某些实施方式中,一个抗体具有从1至8个,或者从2至8个,或者从2至4个工程化的半胱氨酸残基。
非抗体蛋白质
本领域技术人员熟知,尽管这里描述的过程以抗体作为示例,它也可以成功应用于任何具有非配对、在表达或者生产的过程中被硫醇封端的、工程化的或者天然的半胱氨酸残基(在蛋白质上通常不形成链间或者链内键的半胱氨酸)的蛋白质。所述方法特别有帮助的蛋白质是这样的蛋白质,所述蛋白质除了包含非配对的半胱氨酸残基,还包含天然的半胱氨酸残基并形成链间二硫键,特别是那些可以被切断并不立刻导致蛋白去折叠的键。当涉及到非抗体蛋白质时,术语“链间二硫键”是指相邻肽链上的2个半胱氨酸残基之间的共价键。候选的非抗体蛋白质包括:那些具有暴露于溶剂的二硫键,且所述二硫键在天然折叠构象中的稳定性与那些被封端的硫醇的稳定性具有可比性。如本文所用,工程化的半胱氨酸蛋白质,是指选定的氨基酸被突变为半胱氨酸的蛋白质。代表性的蛋白质还包括Fc-融合蛋白,例如包含一个抗体的Fc区域的蛋白质,其共价连接于提供针对目标靶点的特异性的蛋白质。
Ⅳ.实施例
实施例1:实施选择性还原的代表性方法
一个反应容器与一切向流过滤(TFF)设备相连接,并且安装超滤膜(例如,88cm2,Millipore Pelicon 3,再生纤维素)。(许多不同类型的具有合适大小的膜可以应用于上述过程。对于还原剂添加速率计算所需要的流出速率和筛分因素,应该根据方法中采用的膜的渗透通量和膜的类型以及表面积来确定。)根据制造商的说明,对膜进行冲洗和平衡。设置渗滤缓冲液池,它含有缓冲剂(例如,50mM Tris/5mM EDTA,pH 8.0)。它是通过管道与反应容器相连,并且液体流经管道是通过蠕动泵加以控制(例如渗滤缓冲泵)。包含封端的工程化半胱氨酸残基的mAb被放置在反应容器中。反应在室温下进行,反应混合物被连续泵送通过超滤膜,控制滞留线以保证跨膜压力为约20psi。
包含还原剂的溶液池通过管道连接于渗滤缓冲液管线,或者连接于流路或者反应容器的其它位置。计算流速,在该流速下包含还原剂的原液被添加到反应混合物中以维持反应混合物中起始的(或所需要的)还原剂浓度(反应混合物中最优的还原剂浓度采用本文描述的方法并通过实验确定,并且对不同的硫醇是不同的,因为它们具有不同的还原能力,如图1所示)。还原剂浓度在整个反应时间基本上保持不变。渗滤缓冲液也以可控的速率被泵送到反应容器中,以便整个反应体积保持不变(即在恒体积渗滤中),即,还原剂和渗滤缓冲液被引入反应器的速率与通过渗透所导致的体积损失速率相匹配。
实施例2采用半胱氨酸、N-乙酰基半胱氨酸和半胱胺作为还原剂,并在不移除封端副产物的情况下,对工程化的半胱氨酸残基进行脱封端
将S239C工程化的半胱氨酸抗体(IgG1抗体,其在239位为工程化的半胱氨酸残基,根据EU索引进行编号),20mg(135nmol,15mg/mL),用33μL 100mM(3.3μmol)半胱氨酸、N-乙酰基半胱氨酸或者半胱胺,在pH 8.0和室温下处理8h。按每1h间隔取出样品,纯化,与过量SGD-1269偶联,并冷冻储存。纯化和偶联终止了还原反应,保存了在还原过程中产生的二硫键。偶联的样品在变性条件下通过反相HPLC(“rPLRP”)进行分析,由此可以推导出工程化的半胱氨酸残基的脱封端程度(从H0转换为H1)。链间二硫键的断裂程度也可以通过具有偶联有>1mcMMAF(mcMMAF是连接于药物单甲基奥瑞他汀F的马来酰亚胺己酸接头)的重链量加以确定。没有观察到偶联有>2mcMMAF分子的重链。静态溶液还原实验中得出的结果见图1。结果显示,三种硫醇中每一种都能够与ecmAb反应,将工程化的半胱氨酸残基脱封端,但是三者表现很不同。NAC的表现形式可用流程1中的第一个反应最直接地加以描述:反应进行直至富集足够浓度的二硫键产物4,然后因为正反应和逆反应以相同的速率发生而发生停止。半胱氨酸是比NAC更强大的还原剂,但是,与停止在部分活化ecmAb不同,反应发生逆转,再产生ecmAb。这一逆转表明,采用半胱氨酸,会涉及额外的反应,即还原剂的自氧化。
2R-SH+O2→R-S-S-R (iii)
因此,半胱氨酸的自氧化所产生的胱氨酸,也会将活化的工程化的半胱氨酸残基再封端,从而逆转最初的还原。对图1进行仔细查阅,可显示出,采用半胱胺时,也发生了同样的现象(即起始还原后伴随着再封端),但是半胱胺是一种更弱的还原剂,因此起始还原程度没有采用半胱氨酸或者N-乙酰半胱氨酸时高。
实施例3选择性活化ecmAbs上的工程化的半胱氨酸残基
反应容器与一切向流过滤(TFF)设备相连接,并且安装超滤膜(例如,88cm2,Millipore Pelicon 3,再生纤维素)。根据制造商的说明书,对膜进行冲洗和平衡。设置渗滤缓冲液池,它含有缓冲剂(例如,50mM Tris/5mM EDTA,pH 8.0)。它是通过管道与反应容器相连,并且液体流经管道通过蠕动泵加以控制(例如渗滤缓冲泵)。包含封端工程化的半胱氨酸残基的mAb(h2H12S239C ecMab,h1F6 239ecMab,h2H12K326C ecMab或h2H12A327C ecMab,根据EU索引进行编号)被放置在反应容器中,浓度大约为15mg/mL并且pH被调至8.0.。反应在室温下进行,反应混合物被连续泵送通过超滤膜,控制滞留线以保证跨膜压力为约20psi。
包含半胱氨酸的溶液池通过管道与渗滤缓冲液管线连接。计算流速,在该流速下,将包含半胱氨酸的原液添加到反应混合物中以维持反应混合物中起始的(或所需要的)半胱氨酸浓度。如下计算注入到反应混合物中的半胱氨酸原液浓度和添加速率,以便于半胱氨酸浓度在整个反应时间基本上保持不变。在图2-5所示的例子中,半胱氨酸的反应浓度范围为0.5-0.9mM(如图中所描述的说明);半胱氨酸原液浓度为100mM,10mM,或者5mM,并且半胱氨酸被泵送到反应器中的流速也被调节以根据下述化学式维持实验中的半胱氨酸浓度。半胱氨酸的筛分因素经测定是0.8-0.9。在整个实验中,反应混合物中半胱氨酸的浓度采用DTNB分析法周期性地测定,以保证半胱氨酸水平维持在需要的浓度(未显示)。
渗滤缓冲液也以可控的速率被泵送到反应容器中,以便整个反应体积保持不变(即在恒体积渗滤中),即,将半胱氨酸和渗滤缓冲液引入反应器的速率与通过渗透所导致的体积损失速率是相匹配的。
样品在图中所示的时间间隔被移出,在PD-10柱中洗脱纯化,并且与SGD-1269偶联。偶联的样品然后通过rPLRP进行分析。rPLRP图谱分析提供了维持封端(%H0)的重链比例,工程化的半胱氨酸残基被选择性地还原(%H1)的比例,或者工程化的半胱氨酸位点并且额外地在链间二硫键位置被还原(%H2;非选择性还原)的比例。
半胱氨酸添加速率计算
半胱氨酸被添加到反应混合物中的速率与半胱氨酸通过渗透所损失的速率相同。分析物通过渗透所导致初始损失速率可用方程式2计算,这可以来自恒定体积速率的清除率理论方程(即方程式1)推导出。半胱氨酸原液被泵送到反应混合物中以维持起始半胱氨酸浓度的速率见方程式3。
方程式1:C/C0=e(-NS)。C是在任一时间t的浓度;C0是初始浓度;N是时间t时的置换体积数,并且S是分析物的筛分因子(经验值)。置换体积数N,是r*t/Vd,其中r是渗透流速,Vd是批量体积。进行替换,求导,并且评估在t=0时的导数,得到方程式2。
方程式2:dC/dt=-C0*(r/Vd)*S,:其中,dC/dt是半胱氨酸在t0时的损失速率。
在反应混合物中添加半胱氨酸溶液以实现给定浓度的速率见方程式3。
方程式3:R=-dC/dt*Vd/[Cys],其中[Cys]是半胱氨酸的原液浓度。
因此,替换方程式2中的表达式dC/dt,给出了用于添加速率的方程式4,
方程式4:R=C0*r*S/[Cys]=(0.8mM)*(720mL/hr)*(0.8)/(100mM)=4.61mL/hr。
结果
通过在反应过程中连续渗滤ecmAb,并在全过程中将半胱氨酸加入到渗滤缓冲液中,可以实现选择性活化ecmAb上的工程化的半胱氨酸残基。连续渗滤清除了反应副产物。半胱氨酸的添加速率,基于半胱氨酸通过渗滤的理论中半胱氨酸的理论清除率来计算,以便为反应混合物中提供恒定的半胱氨酸浓度。图2显示4.5hr后,约80%可用的工程化的半胱氨酸残基被活化(“%H1”),而链间二硫键的断裂几乎可以忽略(由%H2表示)。这一实验采用0.6mM半胱氨酸在更长的时间下进行重复,得到了类似的结果(图3)。在更高的浓度下,反应更快但是特异性较低。应理解,在整个过程中,反应混合物处于流动状态,伴随半胱氨酸溶液通过半胱氨酸添加泵进入反应混合物,以及通过TFF膜(渗透)离开反应混合物,因此,在整个反应过程中,浓度可以稍微波动,但是活化条件通常可以接近标称浓度。1.5-2mM或者更高浓度的半胱氨酸,会导致链间二硫键断裂增加。图4显示了工程化的半胱氨酸残基活化可以在许多位点选择性地获得,而与抗体的种类无关。K326突变体的活化时间曲线表明,这一位点的活化比S239或A327位点快。这很可能是由于以下事实:K326是更加溶液可及的,但尽管如此,该方法仍获得了理想的选择性活化。图5显示通过采用低浓度的还原剂并在较长时间下反应,可以使得反应被驱动到工程化的半胱氨酸残基被近乎100%活化,并且具有很好的选择性。
尽管为了清楚和理解的目的,通过阐述和例子,对上述内容进行了详细说明,本领域技术人员应理解,在所附权利要求的范围内,可以进行某些改变和变动。此外,在本文中所提供的每个文献,都同样程度地通过引用被整体引入本文,就好比每个文献被单独地引用而引入本文。
- 还没有人留言评论。精彩留言会获得点赞!