纳米粒子药物结合物的制作方法
本申请案分别主张在2014年5月29日和2014年12月19日申请的美国临时专利申请案第62/004,738号和第62/094,923号的优先权以及权利,且其全文通过引用并入本文中。
技术领域
本发明大体上涉及用于传递治疗剂(例如,靶向药物释放)的纳米粒子结合物,所述治疗剂用于检测、预防和治疗癌症以及其它疾病。
背景技术:
纳米治疗传递媒剂通常为尺寸在1到1,000nm范围内的巨或超分子多组分系统,其本身具有治疗性(例如,无活性药物成分)或充当治疗性传递系统。迄今为止,脂质体纳米粒子和生物制剂包含大部分数量的经美国食品药物管理局(FDA)批准的产品或临床试验中的产品,其用于治疗各种癌症类型,而许多基于聚合物的粒子制剂目前正处于早期试验。
用于纳米治疗传递系统的所要理想对象具有共同特征:以控制方式并入和释放药物化合物,其可在使偏离目标毒性降到最低时,有利地改变药物生物可用性和药物动力学。理想地,将成像标记并入其中,以评估其精确定位和在疾病部位的保留。
但是,此等系统使用不同机制起作用。例如,抗体药物结合物(ADC)主要通过主动靶向肿瘤细胞和条件性释放药物分子来达成较低药物毒性。当结合细胞表面抗原时,在细胞内化和内体吸收后出现活性药物释放。另一方面,相较于被动地负载有更大有效负载(阿霉素(Doxil)为约10,000药物分子量)的组合复合物(直径为约20到150nm),通常大得多的脂质体和基于聚合物的药物传递系统已普遍缺乏靶向能力(BIND-014除外)。因此,此等复合物主要依赖于公认增强的渗透和保留(EPR)作用来成功传递纳米调配药物。尽管因为脂质体的尺寸其填隙式渗透可能较差,但通过各种未完全理解的机制来释放自由药物。例如,蛋白结合型紫杉醇(Abraxane,约140nm)依赖于不同方法,以增强疏水性化合物的生物可用性。在此情况下,清蛋白和药物(太平洋紫杉醇)的特定制剂形成初始复合物,反过来当注入时估计其分散成更小蛋白药物凝集物。
因此,需要一种用于药物传递的独特平台,其提供足够生物稳定性,且显示生物活性化合物在所要部位的控制释放。
技术实现要素:
本文呈现纳米粒子药物结合物(NDC)的方法和组合物,具体地说,呈现共价连接有药物分子的基于二氧化硅的纳米粒子平台。NDC已展现为纳米治疗剂。尺寸、分子组成和化学方法(例如,药物释放模式)的组合可充分利用见于其它纳米治疗产品中的有利特性,以便克服阻碍传统制剂的关键障碍物,包括狭小治疗性指数、剂量限制毒性和有限临床效用。
一方面,本发明涉及一种纳米粒子药物结合物(NDC),其包含:纳米粒子(例如,直径在1nm到25nm范围内);键联基团部分;和药物部分(例如,达沙替尼(dasatinib)或吉非替尼(gefitinib),包括其任何类似物),其中纳米粒子包覆有有机聚合物(例如,其中有机聚合物包含至少一个双官能化顺丁烯二酰亚胺硅烷基-聚乙二醇基团,其连接到至少一个键联基团-药物构筑体上),且其中药物部分和键联基团部分形成可裂解(例如,经由蛋白酶)键联基团-药物构筑体,其共价键联到纳米粒子上(例如,通过键联基团部分)(例如,其中药物部分与纳米粒子的平均比率在1到20范围内)。
在某些实施例中,键联基团部分包含一或多种氨基酸(例如,肽或多肽)(例如,1到10种氨基酸)。在某些实施例中,键联基团部分包含(氨基-(间隔基)x)y-肽或(间隔基)z-肽[例如,二肽(例如,苯丙氨酸-精氨酸(Phe-Arg)或苯丙氨酸-赖氨酸(Phe-Lys))],其中间隔基具有2到50个原子(例如,其中间隔基为PEG),其中x为整数1到5,其中y为整数1到5,其中z为整数5到15,且其中键联基团部分在键联基团部分与药物部分之间包含可降解部分(例如,酰胺键)(例如,在蛋白酶存在下使药物部分裂解)。在某些实施例中,键联基团部分在肽与药物部分之间包含间隔基(例如,聚乙二醇(PEG)、PEG2、氨基甲酸对氨基苄氧酯(PABC))。在某些实施例中,NDC进一步包含荧光化合物(例如,与纳米粒子相连,例如,在纳米粒子的核心内)。在某些实施例中,NDC进一步包含放射性标记。
在某些实施例中,键联基团部分能够在蛋白酶(例如,丝氨酸蛋白酶(例如,胰蛋白酶)、半胱氨酸蛋白酶(例如,组织蛋白酶B))结合时,在C端进行水解,由此自纳米粒子释放药物部分。
在某些实施例中,药物部分包含受体酪氨酸激酶(RTK)抑制剂(例如,达沙替尼或吉非替尼,包括其任何类似物(例如,其任何药物和/或治疗性等效物),其经改质以在不扰乱药物部分活性结合位点的基础化学结构的情况下,连接到键联基团部分)。
在某些实施例中,NDC进一步包含1到20种靶向部分(例如,环状精氨酰基甘氨酰基天冬氨酸(cRGD)),其中靶向部分结合到肿瘤细胞上的受体上。
在某些实施例中,NDC为诊疗一体化的。
在某些实施例中,荧光化合物为Cy5.5。
在某些实施例中,将药物部分连接到放射性标记上。
在某些实施例中,纳米粒子进一步包含基于二氧化硅的核心和包围至少一部分核心的二氧化硅外壳。
关于本发明的给定方面描述的实施例的要素可用于本发明另一方面的各种实施例中。例如,在此考虑了,取决于一个独立权利要求的附属权利要求项的特征可用于任何其它独立权利要求的设备和/或方法中。
附图说明
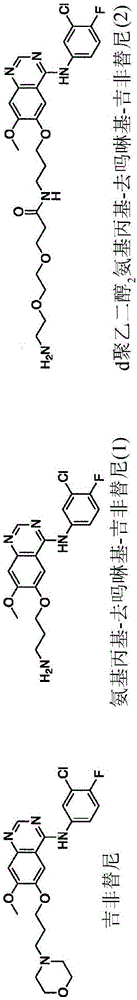
图1A描绘吉非替尼(gefitinib)和类似物(APdMG 1和dPEG2APdMG 2)的化学结构。
图1B描绘通过酰胺键直接连接的键联基团-药物(Phe-Arg-APdMG 3)的化学结构。
图1C描绘通过dPEG2间隔基连接的键联基团-药物(Phe-Arg-dPEG2APdMG 4)的化学结构。
图1D描绘通过可降解PABC间隔基连接的键联基团-药物(Phe-Lys-PABC-APdMG 5)的化学结构。
图2A到2C显示键联基团类型。
图2A描绘Phe-Arg-APdMG将酰胺键用于药物连接。酶类识别出且结合二肽序列(Phe-Arg),随后水解二肽C端的酰胺键,且释放APdMG 1。
图2B显示Phe-Arg-dPEG2APdMG使用dPEG2APdMG 2,其在药物和二肽之间并入10个原子的较长PEG间隔基,以增强药物释放。
图2C显示Phe-Lys-PABC-APdMG在二肽(Phe-Lys)和氨基丙基-dMG之间使用氨基甲酸对氨基苄酰氧酯(或PABC)间隔基团。在经酶催化释放间隔基-药物后,间隔基自药物中自发地分解。
图3A到3C为自键联基团-药物构筑体中经酶(胰蛋白酶)催化的代表性药物释放。数据表明APdMG和dPEG2APdMG自构筑体中释放。在括弧中表明保留时间。在10mM磷酸盐缓冲液(7.2pH值)中,在37℃下进行胰蛋白酶试验。
图3A显示Phe-Arg-APdMG 3(顶部)和60min的Phe-Arg-APdMG+胰蛋白酶(底部)的LCMS数据。
图3B显示Phe-Arg-dPEG2-APdMG 4(顶部)和10min的Phe-Arg-dPEG2-APdMG+胰蛋白酶(底部)的LCMS数据。
图3C显示Phe-Arg-PABC-APdMG 5(顶部)和10min的Phe-Arg-PABC-APdMG+胰蛋白酶(底部)的LCMS数据。
图4A和4B显示自由键联基团-药物构筑体Phe-Arg-APdMG、Phe-Arg-dPEG2APdMG和Phe-Lys-PABC-APdMG的活体外药物释放试验,其通过HPLC在348nm处随时间推移而监测。自由药物%为释放的药物除以在348nm处测定的初始键联基团-药物构筑体的药物负荷。
图4A描绘用胰蛋白酶处理的自由键联基团-药物构筑体。在10mM磷酸盐缓冲液(7.2pH值)中,在37℃下进行胰蛋白酶试验。
图4B描绘用组织蛋白酶B处理的自由键联基团-药物构筑体。在25mM乙酸钠缓冲液(5.0pH值)中进行组织蛋白酶B试验。
图5A和5B显示在胰蛋白酶存在下自NDC的活体外药物释放的代表性HPLC图谱。用胰蛋白酶处理NDC,随后在5和120min后通过HPLC进行分析。数据表明化合物2或3自碳点(C'dots)释放。在10mM磷酸盐缓冲液(7.2pH值)中,在37℃下进行胰蛋白酶试验。在348nm处进行HPLC分析。
图5A显示NDC 6的HPLC图谱。
图5B显示NDC 7的HPLC图谱。
图6A和6B描绘在酶类存在下自NDC的活体外药物释放。通过HPLC在348nm处随时间推移监测酶反应。在10mM磷酸盐缓冲液(7.2pH值)中,在37℃下进行胰蛋白酶试验;在25mM乙酸钠缓冲液(5.0pH值)中,在37℃下进行组织蛋白酶B试验。
图6A显示用胰蛋白酶处理的碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG 6的药物释放。
图6B显示用组织蛋白酶B处理的碳点-(Cy5)-PEG-Phe-Lys-PABC-APdM 7的药物释放。
图7显示用吉非替尼、碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG 6和碳点-(Cy5)-PEG-Phe-Lys-PABC-APdMG 7处理的H1650细胞的西方墨点分析。在指定浓度下用吉非替尼或指定NDC处理细胞达18小时,随后用EGF(50ng/mL)处理达5分钟(pEGFR-磷酸化EGFR;tEGFR-总EGFR)。
图8显示碳点-(Cy5)-PEG-Phe-Arg-dPEG2-Gly-D-Tyr(131I)-APdMG 8的放射性GPC。基于峰积分>90%的放射性化学产率。推测更小峰为残余自由131I。
图9A和9B显示键联基团-药物构筑体Phe-Arg-dPEG2-D-Tyr-氨基丙基-dMG和Phe-Lys-PABC-D-Tyr-氨基丙基-dMG,其将d-酪氨酸残基与药物组分合并以连接放射性标记(化合物23和24)。
图10显示流程1,其说明自纳米粒子药物结合物(NDC)的酶介导的药物释放。
图11显示流程2,其说明碳点-(Cy5)-PEG-mal与键联基团-药物构筑体Phe-Arg-dPEG2APdMG 4和Phe-Lys-PABC-APdMG 5反应,产生NDC碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG 6和碳点-(Cy5)-PEG-Phe-Lys-PABC-APdMG 7。
图12显示流程3,其说明APdMG 1的合成过程。
图13显示流程4,其说明dPEG2APdMG 2的合成过程。
图14显示流程5,其说明Phe-Arg-APdMG 3和Phe-Arg-dPEG2APdMG 4的合成过程。
图15显示流程6,其说明Phe-Lys-PABC-APdMG(5)的合成过程。
图16A到16D显示mal-PEG-碳点以及NDC 6和7的特性描述。
图16A显示在348nm处的分析性C18反相HPLC。
图16B显示TEM图像。
图16C显示吸光度和发射光谱。
图16D显示FCS相关性曲线。
本发明的特征和优点将自以下在结合图式时所阐述的详细描述变得更显而易见,其中相似参考符号始终标识对应要素。在图式中,相似参考标号通常表明相同、功能上相似和/或结构上相似的要素。
定义
为了使本发明更容易理解,在下文首先对某些术语进行定义。在整个说明书中阐述以下术语和其它术语的其它定义。
在本申请案中,除非另外说明,否则使用“或”意味着“和/或”。如本申请案中所使用,术语“包含(comprise)”和所述术语的变体,诸如“包含(comprising、comprises)”并不意欲排除其它添加剂、组分、整数或步骤。如本申请案中所使用,术语“约”和“大约”用作等效物。在存在或不存在约/大约的情况下,用于本申请案中的任何数值皆意味着涵盖相关领域中普通技术人员所了解的任何正常波动。
在某些实施例中,除非另行说明或另外自上下文显而易见,否则术语“大约”或“约”是指落入所述参考值的任一方向(大于或小于)25%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%或更小的一系列值(除了其中此等数值将超过100%的可能值)。
术语“投与”是指将物质引入至个体中。一般来说,可使用任何投与途径,包括例如不经肠(例如,静脉内)、口服、局部、皮下、经腹膜、动脉内、吸入、经阴道、经直肠、经鼻、引入至脑脊髓液中或滴注至身体区室中。在一些实施例中,投与为口服。另外或或者,在一些实施例中,投与为不经肠投与。在一些实施例中,投与为静脉内投与。
术语“试剂”是指任何化学类别的化合物或实体,包括例如多肽、核酸、醣类、脂质、小分子、金属或其组合。如将自上下文而明确,在一些实施例中,试剂可为或包含细胞或生物体、或其部分、提取物或组分。在一些实施例中,因为试剂在自然中发现和/或从自然中获得,所以试剂为或包含天然产物。在一些实施例中,因为试剂是通过人手的行为所设计、工程化和/或制造,且/或并非在自然中发现,所以试剂为或包含一或多种人造实体。在一些实施例中,可以分离或纯净形式使用试剂;在一些实施例中,可以粗产物形式使用试剂。在一些实施例中,提供潜在试剂作为集合或库,例如可对其进行筛查以识别或特性化其中的活性剂。一些可使用的试剂的具体实施例包括小分子、抗体、抗体片段、适体、siRNA、shRNA、DNA/RNA混合物、反义寡核苷酸、核酶、肽、肽模拟物、肽核酸、小分子等。在一些实施例中,试剂为或包含聚合物。在一些实施例中,试剂含有至少一种聚合部分。在一些实施例中,试剂包含治疗剂、诊断剂和/或药物。
术语“肽”或“多肽”是指通过肽键键联在一起的至少两种(例如,至少三种)氨基酸的链带。在一些实施例中,多肽包含天然产生的氨基酸;或者或另外,在一些实施例中,多肽包含一或多种非天然氨基酸(即,自然界中不存在但可并入到多肽链中的化合物;参见,例如http://www.cco.caltech.edu/~dadgrp/Unnatstruct.gif,其显示已成功并入到官能离子通道中的非天然氨基酸的结构),和/或可作为替代方案使用的本领域中已知的氨基酸类似物)。在一些实施例中,蛋白质中的氨基酸中的一或多者可例如通过添加诸如碳水化合物基团、磷酸基、法呢基、异法呢基、脂肪酸基团、用于结合的键联基团、官能化或其它改质等进行改质。
如本文所使用,术语“相连”通常是指两种或多于两种实体彼此在物理上直接或间接地接近(例如,经由充当键联基团的一或多种其它实体),以形成足够稳定的结构,以使实体在相关条件,例如生理条件下保持物理上接近。在一些实施例中,相连部分彼此共价键联。在一些实施例中,相连实体为非共价键联。在一些实施例中,相连实体通过特定非共价相互作用彼此键联(即,通过相互作用配位体之间的相互作用,其辨别其相互作用搭配物和使用上下文中所存在的其它实体,诸如抗生蛋白链菌素/抗生物素蛋白相互作用、抗体/抗原相互作用等)。或者或另外,足够数量的较弱非共价相互作用可为部分提供足够稳定性以保持相连。例示性非共价相互作用包括(但不限于)静电相互作用、氢键结、亲和力、金属配位、物理吸附、主体-客体相互作用、疏水性相互作用、π堆叠相互作用、凡得瓦尔力(van der Waals)相互作用、磁性相互作用、静电相互作用、偶极-偶极相互作用等。
如本文所使用,“生物可降解”材料为当引入至细胞中时,通过细胞机制(例如,酶降解)分解或通过水解成为细胞可再使用或处理的组分,且不对细胞造成显著毒性作用的那些材料。在某些实施例中,由生物可降解材料分解所产生的组分不会在活体内诱发发炎和/或其它不良作用。在一些实施例中,生物可降解材料以酶促方式分解。或者或另外,在一些实施例中,生物可降解材料通过水解分解。在一些实施例中,生物可降解聚合材料分解成为其组份聚合物。在一些实施例中,生物可降解材料(包括例如生物可降解聚合材料)的分解包括酯键的水解。在一些实施例中,材料(包括例如生物可降解聚合材料)的分解包括氨基甲酸酯键的裂解。
如本文所使用,“官能性”生物分子为呈显示其特性化特性和/或活性形式的生物分子。生物分子可具有两种官能团(即,双官能)或许多官能团(即,多官能)。
如本文所使用,术语“活体外”是指在人工环境,例如在试管或反应器中、在细胞培养液中等,而非在多细胞生物体内发生的现象。
如本文所使用,“活体内”是指在多细胞生物体,诸如人类和非人类动物内发生的现象。在基于细胞的系统的情况下,所述术语可用于指代在活细胞(与例如活体外系统相反)内发生的现象。
如本文所使用,术语“成像剂”是指任何元素、分子、官能团、化合物、其片段或部分,其有助于检测与其连接的试剂(例如,多醣纳米粒子)。成像剂的实例包括(但不限于):各种配位体、放射性核素(例如,3H、14C、18F、19F、32P、35S、135I、125I、123I、64Cu、187Re、111In、90Y、99mTc、177Lu、89Zr等)、荧光染料(针对特定例示性荧光染料,参见下文)、化学发光剂(诸如吖啶酯、稳定化二氧杂环丁烷及类似者)、生物发光剂、光谱可解析无机荧光半导体纳米晶体(即,量子点)、金属纳米粒子(例如,金、银、铜、铂等)纳米簇、顺磁金属离子、酶类(针对酶类的特定实例,参见下文)、色度标记(诸如染料、胶态金及类似者)、生物素、地高辛(digoxigenin)、半抗原和抗血清或单株抗体可适用的蛋白质。
如本文所使用,术语“纳米粒子”是指直径小于1000纳米(nm)的粒子。在一些实施例中,如由美国国家科学基金会(National Science Foundation)定义,纳米粒子具有小于300nm的直径。在一些实施例中,如由美国国立卫生研究院(National Institutes of Health)定义,纳米粒子具有小于100nm的直径。在一些实施例中,纳米粒子为微胞,因为其包含通过微胞膜自主体溶液分离的闭合区室,其通常包括包围且围封空间或区室的两亲实体(例如,以定义内腔)。在一些实施例中,微胞膜包括至少一种聚合物,诸如生物兼容性和/或生物可降解聚合物。
如本文所使用,术语“个体”包括人类和哺乳动物(例如,小鼠、大鼠、猪、猫、狗和马)。在许多实施例中,个体为哺乳动物,尤其是灵长类动物,尤其是人类。在一些实施例中,个体为家畜,诸如牛、绵羊、山羊、奶牛、猪及类似者;家禽,诸如鸡、鸭、鹅、火鸡及类似者;和家养动物,尤其是宠物,诸如狗和猫。在一些实施例中(例如,尤其在研究背景中),个体哺乳动物将为例如啮齿动物(例如,小鼠、大鼠、仓鼠)、兔、灵长类动物或猪,诸如近交猪及类似者。
如本文所使用,术语“治疗(treatment)”(亦为“治疗(treat)”或“治疗(treating)”)是指物质的任何投与,其部分或完全地缓解、改善、减轻、抑制一或多种症状、特征和/或具体疾病、病症和/或病况的病因、延缓其发作、降低其严重性和/或减小其发病率。此类治疗可为对并未显示相关疾病、病症和/或病况的征象的个体的治疗,和/或对仅显示疾病、病症和/或病况的早期征象的个体的治疗。或者或另外,此类治疗可为对显示相关疾病、病症和/或病况的一或多种确定征象的个体的治疗。在一些实施例中,治疗可为对已诊断为患有相关疾病、病症和/或病况的个体的治疗。在一些实施例中,治疗可为对已知具有一或多种在统计学上与相关疾病、病症和/或病况发展风险增加相关的易感性因素的个体的治疗。
具体实施方式
本文描述纳米粒子药物结合物(NDC),其在某些实施例中包含具有共价连接的药物分子/部分的无毒、多形态、经临床证实的基于二氧化硅的纳米粒子平台。纳米粒子药物结合物(NDC)显示出成像能力和通过肾脏有效清除的靶向配位体。此外,结合物并入治疗剂用于癌症检测、预防和/或治疗。例如,已合成含有特定受体酪氨酸激酶(RTK)抑制剂的NDC,且其展现以控制和可预测方式释放药物化合物。此外,西方墨点分析显示细胞中RTK磷酸化水平降低,表明基于NDC的活体外药物传递。
在一些实施例中,基于二氧化硅的纳米粒子平台包含超小纳米粒子或“碳点”,其是可控制直径低至10nm范围、具有一系列模块化官能团的荧光、有机二氧化硅核-壳型粒子。碳点由美国专利第8298677B2号“基于二氧化硅的荧光纳米粒子(Fluorescent silica-based nanoparticles)”、美国公开案第2013/0039848A1号“基于二氧化硅的荧光纳米粒子(Fluorescent silica-based nanoparticles)”和美国公开案第US 2014/0248210 A1号“基于二氧化硅的多模态纳米粒子(Multimodal silica-based nanoparticles)”来描述,所述专利的内容以全文引用的方式并入本文中。并入到核心的二氧化硅基质中的是近红外染料分子,诸如Cy5.5,其提供其独特的光学特性。包围核心的是二氧化硅层或二氧化硅壳。二氧化硅表面用硅烷基-聚乙二醇(PEG)基团进行改质,以增强在水性和生物学上相关的条件中的稳定性。已对此等粒子进行活体内评估,且主要由于其尺寸和惰性表面,其显示出极佳清除特性。并入到碳点中的其它官能性为化学感测、非光学(PET)图像对比度和活体外/活体内靶向能力,使其能够用于可视化淋巴结用于手术应用和癌症中的黑素瘤检测中。
碳点因其物理特性以及展现出的人类活体内特性而为药物传递提供独特平台。在保持所要清除和药物动力学特性的同时,此等粒子为超小的,且受益于肿瘤微环境中的EPR效应。为此目的,本文描述一种纳米粒子药物传递系统,其中在某些实施例中,药物构筑体共价连接到碳点(或其它纳米粒子)。用于药物传递的基于碳点的NDC提供良好生物稳定性、使过早药物释放降至最低且显示出生物活性化合物的控制释放。在某些实施例中,基于肽的键联基团用于NDC应用。这些键联基团在抗体和聚合物的情况下,活体外和活体内皆为稳定的,其具有依赖于通过溶酶体蛋白酶进行酶催化水解的高度可预测释放动力学。例如,可使用组织蛋白酶B(一种在溶酶体中高度表现的蛋白酶)以促进药物自大分子释放。通过在大分子主链与药物分子之间并入短、对蛋白酶敏感的肽,可在酶存在下获得药物的控制释放。
在某些实施例中,NDC为超小的(例如,平均直径为约5nm到约10nm,(例如,约6nm))且使用对酶敏感的键联基团,例如其中通过蛋白酶催化药物释放。在一个例子中,对吉非替尼,一种重要表皮生长因子受体突变体(EGFRmt+)-酪氨酸激酶抑制剂(TKI)癌症药物,进行改质且将其并入到粒子上。所得NDC显示出极佳活体外稳定性、可溶性,且被证明在EGFRmt+-表现NSCLC细胞中为活性的。
在某些实施例中,NDC包含一或多种靶向部分,例如以瞄准特定组织类型(例如,特定肿瘤)。具有靶向部分的NDC增强肿瘤细胞中药物的内化(例如,靶向配位体结合到肿瘤细胞上的受体上,和/或将药物传送到肿瘤细胞中(例如,通过提高的渗透性))。例如,为了制造具有其它靶向部分的粒子治疗剂(例如,cRGD),将二氧化硅纳米粒子添加到cRGDY-PEG结合物与顺丁烯二酰亚胺双官能化PEG的混合物中。顺丁烯二酰亚胺双官能化PEG支持药物-键联基团结合物的其它连接,以制造诊疗一体化产品。
在一些实施例中,超小粒子可与PET标记和/或光学探针相连。可在活体内观测纳米粒子(例如,经由PET),以评估靶向部位中的药物累积。例如,可首先投与具有PET标记(例如,在无药物物质的情况下)的纳米粒子。随后,通过分析纳米粒子的活体内PET图像,可估算肿瘤中的药物(例如,结合有纳米粒子)浓度和累积率。基于所获得的估算量可确定剂量,以提供个体化用药(例如,肿瘤尺寸而非患者体重)。在一些实施例中,可对受到放射性标记的药物进行活体内追踪。如果未靶向高度浓缩的化学治疗药物,那么所述药物具有潜在危险。在一些实施例中,具有光学探针的纳米粒子(例如,荧光团)可用于手术中成像(例如,当暴露组织/肿瘤的表面时)和/或肿瘤的活组织检查。
可独立地对治疗剂和纳米粒子进行放射性标记或光学标记,从而允许独立监测治疗剂和纳米粒子。在一个实施例中,放射性氟化(即,18F)达沙替尼(dasatinib)经由NHS酯键与连接到纳米粒子上的PEG-3400部分偶合。放射性氟对于能够独立地监测药物自放射性碘标记C24I)荧光(Cy5)纳米粒子的分布和释放中的时间依赖性变化来说为至关重要的。以此方式,可监测前药物(达沙替尼)和纳米粒子。相较于不使用双重标记方法的先前技术中的方法,此准许前药设计的最佳化。在另一实施例中,放射治疗性碘分子(例如,131I)或其它治疗性γ或α发射体经由顺丁烯二酰亚胺官能团与PEG结合,其中治疗剂可能不在活体内自PEG解离。
NDC为通过分子键联基团共价连接到碳点纳米粒子(或其它纳米粒子)的药物化合物。在某些实施例中,键联基团并入对胰蛋白酶(对照酶)和/或组织蛋白酶B敏感的肽(例如,二肽)序列,所述酶主要是在细胞溶酶体中发现的酶。针对控制药物释放,本文中描述了包含两种类别的键联基团化学物质的实验:其一者在键联基团与药物之间并入酰胺键;且另一者在键联基团与药物之间使用可降解部分。在一些实施例中,键联基团经设计以在特定条件,例如蛋白水解下自纳米粒子(例如,碳点)释放药物。
可使用的例示性药物包括RTK抑制剂,诸如达沙替尼和吉非替尼,其可靶向通过人类或鼠类源的原发肿瘤细胞(例如,高级神经胶瘤的经基因工程化小鼠模型、来自人类患者脑部肿瘤外植体的神经球)和/或非神经源的肿瘤细胞系所表现的血小板衍生生长因子受体(PDGFR)或EGFRmt+。可合成达沙替尼和吉非替尼类似物,以在不扰乱界定活性结合位点的基础化学结构的情况下,能够实现与数种键联基团的共价连接。
合成方法得到证实,且获得所要键联基团-药物构筑体和NDC。用于NDC特性描述的HPLC/LCMS方法和酶释放试验也得到发展。活体外酶药物释放试验揭示了NDC设计中的许多重要结构因素。例如,使用不同尺寸的PEG链,碳点与键联基团之间的间隔会不同,且揭示碳点与键联基团之间的足够间隔对允许酶催化药物释放而言为至关重要的。类似地,同样发现键联基团与药物之间的间隔对酶介导的释放而言为至关重要的。此外,相较于那些使用简单酰胺键的键联基团设计,在键联基团和药物之间使用可降解部分的键联基团设计显示出明显更快的释放动力学。在一些实施例中,可降解部分可为碳水化合物和/或任何可以酶促方式裂解和/或活化的键联基团。
还进行了基于细胞的试验。针对原生脑肿瘤细胞(神经球)和/或转移到脑部的肿瘤细胞系(例如,肺、鳞状细胞癌),测试并入有达沙替尼或吉非替尼类似物的键联基团-药物构筑体和NDC。在暴露于NDC以及自由键联基团-药物构筑体的细胞系中,EGFRmt+的磷酸化水平得到降低或消除,且在一些情况下,NDC展现出比天然药物有效的抑制活性。
一方面,本文描述一种纳米粒子药物结合物(NDC),其包含药物部分、键联基团部分和纳米粒子,其中药物部分经由键联基团部分共价键联到纳米粒子上。在某些实施例中,NDC在键联基团部分与药物部分之间包含酰胺键和/或可降解部分。在某些实施例中,键联基团部分包含肽(例如,二肽)。在某些实施例中,键联基团部分在蛋白酶结合时提供C端水解,由此自纳米粒子释放药物部分。在某些实施例中,药物部分包含受体酪氨酸激酶(RTK)抑制剂(例如,达沙替尼或吉非替尼,包括其任何类似物,例如其任何药物和/或治疗性等效物,其经改质以在不扰乱药物部分的活性结合位点的基础化学结构的情况下,连接到键联基团部分)。在某些实施例中,纳米粒子为较新一代碳点。另一方面,本发明针对一种检测、预防和/或治疗疾病(例如,癌症)的方法,其包含投与和/或检测本文所述实施例中任一者的NDC。
纳米尺寸药物传递媒剂之所以吸引人,是因为(1)其尺寸小,能够实现在整个身体中以及在细胞内的运输;(2)其表面积与体积的比率高,能够实现货物负载和释放;以及(3)其具有可调节的表面化学性质,促进可溶性、控制结合并且并入有生物活性官能团。
活体内纳米粒子药物传递充满了大量生物物理学和生物化学挑战,其可造成粒子吸收(调理作用)、排泄(肾脏)或非特异性损失(外渗),且阻止治疗性有效负载到达所要细胞。药物传递构筑体的关键参数之一为其物理尺寸,其中较小粒子(例如,小于或等于约5nm流体动力直径的粒子)可不明确外渗,而大得多的粒子或凝集物(例如,大于或等于约500nm直径的粒子或凝集物)可寄宿在微脉管内,而非经运输到其预定目标。针对非生物可降解材料,发现在限制肾脏清除率以实现所要药物动力学时,优选直径在5nm到10nm范围内,其允许肾脏过滤作为一种粒子去除手段。另外,发现此尺寸范围的粒子还可利用增强的渗透和保留(EPR)作用,即因渗漏脉管导致的肿瘤微环境中的大分子被动累积。
例如在某些实施例中,如美国公开案第2014/0248210A1号所描述,在人体中测试超小(例如,直径在5nm到10nm范围内)粒子,所述公开案的全文通过引用并入本文中。在此实例中,五位患者未产生不良现象,且试剂在研究期间耐受良好。药物动力学行为,其表示为每克组织所注射剂量的百分比(%ID/g)对比注射后时间和对应平均器官吸收剂量,与针对其它常用诊断放射性示踪剂所发现的药物动力学行为相当。此代表性患者的连续PET成像显示假定血池活性自主要器官和组织的进展性损失,到注射后(p.i.)72小时未看到明显的活性。估算此等患者中的全身清除半衰期在13到21小时范围内。有趣的是,与许多疏水性分子、蛋白质和较大粒子平台(大于10nm)相反,在肝脏、脾脏或骨髓中不存在显著的定位。虽然患者经碘化钾(KI)预处理以阻断甲状腺组织吸收,但相对于其它组织,在此患者中获得较高平均吸收甲状腺剂量。粒子同样主要通过肾脏经肾和膀胱壁(在甲状腺和肿瘤后,参见下文)排泄,其展现到注射后72小时最高%ID/g值之一;这常常是通过肾排泄的放射性药物的情况,膀胱壁得到的平均吸收剂量高于其它主要器官和组织。此等发现强调了事实:肾脏而非肝胆排泄是自身体清除的主要途径。
表皮生长因子受体(EGFR)用于靶向疗法。已在10%到35%转移性非小细胞肺癌(NSCLC)中发现导致构成性活化的EGFR突变,且尽管EGFR抑制剂对全身性疾病有效,但药物传递仍限制脑部癌转移的控制。在40%到50%原生多形性胶质母细胞瘤(GBM)中同样发现EGFR突变-两种普遍的脑部癌症形式。尽管EGFR-酪氨酸激酶抑制剂(TKI),诸如吉非替尼,已在临床前环境中显示出前景,但可能由于较差组织或中枢神经系统(CNS)穿透和剂量限制毒性,其已展现在脑部癌症患者中为基本上无效的。
吉非替尼结合且抑制EGFR的激酶结构域活性位点。为了在NDC的情况下使用吉非替尼,至关重要的是并入不会明显干扰药物与激酶结构域结合的化学反应性基团。X射线结晶学和SAR研究揭示,用胺替换吗啉基不会明显改变药物活性,而且为改质和最终与碳点的共价连接提供所需的化学官能团(胺)(图1A)。
实验实例
一个实例展现纳米粒子药物结合物(例如,共价连接有药物分子的基于二氧化硅的纳米粒子平台)的例示性合成和其特性描述以及初步生物评估。
利用去吗啉基-吉非替尼(dMG)的商业可用性,通过对受Boc保护的氨基丙基溴化物进行亲核性取代(例如,在一个步骤中)来获得所要氨基丙基-dMG(APdMG),随后进行酸式去除保护基(图1A、图10(流程1))。另外,通过偶合Fmoc-dPEG2-COOH,随后进行碱式去除保护基步骤,易于自1中获得吉非替尼类似物2,在下文中进一步详细描述所述类似物(图1A、图11(流程2))。为了确保APdMG 1和dPEG2APdMG 2针对EGFR已保留活性,用化合物处理H1650细胞,且通过西方墨点进行分析,以评估EGFR中的磷酸-Tyr168水平。H1650细胞为衍生自人类肿瘤的模型非小细胞肺癌(NSCLC)株(细支气管肺泡癌),其含有导致受体构成性活性的突变EGFR(L858R和ΔE746-A750)。两种化合物均显示出类似于吉非替尼的作用,在1和10μM浓度下抑制磷酸-Tyr168,而dPEG2APdMG 2显示出降低的活性。
针对基于碳点的药物传递,研究了三种键联基团类型(图1B到1D)。三种键联基团类型包括将蛋白酶用于药物释放的二肽序列。蛋白酶识别且结合二肽,导致在C端的水解,自键联基团中释放药物组份。两种模型蛋白酶,胰蛋白酶和组织蛋白酶B,用于评估本文所述的键联基团-药物构筑体。胰蛋白酶选为代表性丝氨酸蛋白酶。其针对含有碱性氨基酸,诸如精氨酸和赖氨酸的肽具有高度活性,且裂解此等残基的C端。组织蛋白酶B为具有较严格的基质特异性的半胱氨酸蛋白酶。迄今为止所述最少基质共同序列为含有疏水性和碱性残基的二肽基序。类似于胰蛋白酶,组织蛋白酶B裂解碱性氨基酸的C端。二肽苯丙氨酸-精氨酸(Phe-Arg)和苯丙氨酸-赖氨酸(Phe-Lys)为胰蛋白酶和组织蛋白酶的胰蛋白酶/组织蛋白酶B识别基序,且包括在键联基团-药物构筑体中(图1B到D)。
Phe-Arg-APdMG为用于获得对键联基团-药物构筑体敏感的蛋白酶的方法的实例(图2A和2B)。在此设计中,将吉非替尼类似物1直接连接到二肽序列的C端。使用固相肽合成(SPPS)方法合成化合物3,随后用2对C端进行改质,且最终进行去除保护基步骤来获得3(图11(流程3))。
考虑到药物组份与二肽基序极为接近,可能阻碍酶结合和水解键联基团-药物的潜在空间问题得到解决。为了增加二肽与药物之间的距离,对APdMG 1进行改质,以获得dPEG2APdMG 2(图13,(流程4))。二肽组份和dPEG2APdMG 2之间进行偶合反应,随后进行去除保护基步骤,得到Phe-Arg-dPEG2APdMG 4,即在Phe-Arg与吉非替尼类似物之间含有10个原子的短链PEG间隔基的键联基团药物构筑体。
为了在不对APdMG 1造成结构改变的情况下保留二肽与药物组分之间增加的间隔,合成Phe-Lys-PABC-APdMG 5。此键联基团在肽和药物之间并入自分解氨基甲酸对氨基苄氧酯(PABC)基团(图1D和2A到2C)。在酶水解时,此基团进一步分解成对氨基苯甲基醇和CO2,由此释放APdMG。以Fmoc-Lys(Mtt)-OH开始化合物5的合成(图13(流程6))。用对氨基苯甲基醇对受保护的氨基酸进行改质,得到Fmoc-Lys(Mtt)-PABA 18。在去除Fmoc基团且与Fmoc-Phe-OH偶合时,形成受保护的二肽Fmoc-Phe-Lys(Mtt)-PABA19。随后用对硝基苯酚碳酸盐氯化物活化-PABA的自由羟基,产生活化碳酸盐20,其随后与APdMG 1反应,产生化合物21。在一轮去除保护基和偶合后,获得化合物22。最终去除保护基步骤需要酸性条件。但是,氨基甲酸对氨基苄氧酯基团本身在此类条件(例如,酸性条件)下容易发生分解。发现足够温和的条件(例如,0.5%TFA)以自赖氨酸中去除Mtt基团且自封端硫醇中去除Mmt基团,同时保留键联基团,以获得所要产物16。掩蔽赖氨酸侧链的Mtt基团较适用于此整体合成方法,因为其在对硝基苯酚碳酸盐氯化物存在下为稳定的,但在温和酸性条件下对去除而言为不稳定的。此与在对硝基苯酚碳酸盐氯化物存在下易于去除的更为常用的超不稳定Mmt基团(其用于赖氨酸侧链保护)形成对比。
为了评估三种键联基团-药物构筑体,对化合物3到5进行酶促水解(表1、图3A到3C、4A和4B。)将药物-键联基团构筑体与胰蛋白酶或组织蛋白酶B一起培育,且通过HPLC或LCMS监测反应。针对所有三种构筑体,胰蛋白酶为活性的:到60min观测到Phe-Arg-APdMG 3的完全水解,其产生APdMG 1;仅到10min,观测到dPEG2APdMG 2自Phe-Arg-dPEG2APdMG 4的完全释放和APdMG 1自Phe-Lys-PABC-APdMG 5的释放。但是,当用组织蛋白酶B处理构筑体时,对于Phe-Arg-APdMG 3观测不到水解,同时Phe-Arg-dPEG2APdMG 4完全水解,导致dPEG2APdMG 2的释放。
下文表1说明通过药物释放试验所获得的键联基团-药物构筑体半衰期。
表1
NH-无水解
a如通过HPLC在348nm处所测定,50%药物自键联基团或粒子中释放的时间。
对化合物3到5进行活体外试验,以获得药物释放图谱,且在不同时间点监测构筑体的酶介导的水解(图4A和4B)。对于Phe-Arg-APdMG 3,50%药物APdMG 1在9min内释放,而Phe-Arg-dPEG2APdMG 4明显更快,在2min内释放50%药物。化合物5同样快速,对于50%药物释放,其需要小于1min。在组织蛋白酶B存在下,因为未观测到药物释放,所以Phe-Arg-APdMG 3经证明为较差基质。对于Phe-Arg-dPEG2APdMG 4,50%药物在110min内释放。对于50%药物,Phe-Lys-PABC-APdMG 5需要<1min,表明其对于酶而言为高效基质。
对于胰蛋白酶和组织蛋白酶B二者,三种键联基团-药物构筑体的药物释放速率遵循相同大体趋势:Phe-Lys-PABC-APdMG 5>Phe-Arg-dPEG2APdMG 4>Phe-Arg-APdMG3(最快到最慢)。Phe-Arg-APdMG 3和Phe-Arg-dPEG2APdMG 4的结果表明药物与二肽单元的接近度影响酶活性和药物释放。当通过併入10原子PEG基团使药物与二肽之间的间隔(距离)增加时,水解(药物释放)得到增强。通过不能够水解构筑体3的组织蛋白酶B,最显著地观测到此作用。但是,通过在药物与二肽4之间并入10原子PEG间隔基,则观测到水解和药物释放。
合成顺丁烯二酰亚胺官能化碳点(碳点-(Cy5)-PEG-mal)来制备NDC。制备用Cy5荧光团改质的硅烷,且用四甲基正硅烷(TMOS)滴定到NH4OH稀释溶液中(摩尔比:TMOS:Cy5:NH3:H2O为1:0.001:0.44:1215),且使其混合达24小时(浦田C(Urata C),青山Y(Aoyama Y),利根川进A(Tonegawa A),山内Y(Yamauchi Y),黑田K(Kuroda K).用于去除表面活性剂以形成胶态间隙孔二氧化硅纳米粒子的透析方法(Dialysis process for the removal of surfactants to form colloidal mesoporous silica nanoparticles).化学通讯(Chem Commun,Camb).2009;(34):5094-6)(山田H(Yamada H),浦田C(Urata C),青山Y(Aoyama Y),长田S(Osada S),山内Y(Yamauchi Y),黑田K(Kuroda K).制备具有不同直径的胶态间隙孔二氧化硅纳米粒子和其在静态水性系统中的独特降解行为(Preparation of Colloidal Mesoporous Silica Nanoparticles with Different Diameters and Their Unique Degradation Behavior in Static Aqueous Systems),化学材料(Chem.Mater).2012;24(8):1462-71.)(王J(Wang J),菅原-鸣泷A(Sugawara-Narutaki A),深尾M(Fukao M),横井T(Yokoi T),下岛A(Shimojima A),大久保T(Okubo T).用胺或氨催化剂两相合成单分散二氧化硅纳米球和其控制自组装(Two-phase synthesis of monodisperse silica nanospheres with amines or ammonia catalyst and their controlled self-assembly).ACS应用材料交界(ACS Appl Mater Interfaces).2011;3(5):1538-44.)。此产生Cy5囊封二氧化硅粒子,通过用PEG-硅烷(500g/mol)(铃木K(Suzuki K),碇K(Ikari K),今井H(Imai H).使用双表面活性剂系统合成具有良序中孔结构的二氧化硅纳米粒子(Synthesis of silica nanoparticles having a well-ordered mesostructured using a double surfactant system).美国化学会志(J Am Chem Soc).2004;126(2):462-3.)和顺丁烯二酰亚胺-PEG-硅烷(摩尔比:PEG-硅烷:TMOS:mal-PEG-硅烷为1:2.3:0.006)处理,用顺丁烯二酰亚胺基团使所述二氧化硅粒子表面进一步聚乙二醇化且官能化。48小时后,通过凝胶过滤来透析、过滤和纯化反应混合物。分别通过荧光相关度光谱测定(FCS)、透射电子显微法(TEM)和直径、形态以及整体纯度的分析性HPLC使纳米粒子特性化。所得碳点直径小于10nm,具有狭窄的粒度分布(图16A到16D)。
通过将Phe-Arg-dPEG2APdMG(4)和Phe-Lys-PABC-APdMG(5)添加到碳点-(Cy5)-PEG-mal中以使构筑体上的封端硫醇与粒子上的顺丁烯二酰亚胺基团反应来获得并入键联基团-药物4和5(例如,碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG(6)和碳点-(Cy5)-PEG-Phe-Lys-PABC-APdMG(7))的NDC(流程2)。遵循通过凝胶过滤纯化来分离NDC产物,且获得NDC 6和7,且通过TEM和HLPC使其特性化(图16A和图16B)。分析性HPLC用于评估污染物的存在以及测定每粒子药物分子的数量或药物与粒子比率(DPR)(表2)。由于Cy5嵌入在碳点内,所以可易于在348nm处测量吉非替尼类似物的浓度,同时可在650nm处获得粒子浓度。尽管平均DPR经证明为适中的,但NDC显示出可测量的不均匀性,其中DPR估计值的范围为小于1到大于15。因为吉非替尼类似物的较差可溶性,所以本可预料到沉淀,但未观测到NDC沉淀。FCS用于评估因键联基团-药物结合而导致的粒度中的变化。如表2中显示,NDC在基质mal-碳点上显示最小直径增加量。
下文表2说明纳米粒子特性描述的概述。
表2
DPR-药物与粒子的比率
a通过FCS测定
b通过HPLC测定
为碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG和碳点-(Cy5)-PEG-Phe-Lys-PABC-APdMG测量随时间推移的酶依赖性药物释放,以获得活体外药物释放图谱(图6A和6B)。图5A和5B中显示代表性HPLC数据,所述数据展现使用胰蛋白酶的药物释放。对于胰蛋白酶而言,NDC 6和7为极佳基质,其分别需要44min和6min以实现50%药物释放(图6A、表1)。在组织蛋白酶B存在下,两种NDC的释放动力学显著减慢:NDC 6在560min内实现50%药物释放,且NDC 7在510min内内实现50%药物释放(图6B、表1)。综合而言,数据展现粒子表面上键联基团-药物构筑体的可获取性,其导致药物组分的控制释放。
在37℃下在酸性和中性pH值(5.0和7.2)下,在含水条件下评估NDC的稳定性。如通过HPLC所测量,NDC 6和7到48小时均显示无降解或药物释放。由于可在活体内发生的可能反向迈克尔(Michael)或硫醇交换反应,观测到自抗体药物结合物的键联基团药物构筑体损失,所以基于硫醇-顺丁烯二酰亚胺的结合引起仔细检查。为了评估在过量硫醇存在下NDC的活体外稳定性,在7.2pH值下在37℃下将NDC 7与30mM谷胱甘肽一起培育达48小时。48小时后,小于5%的键联基团-药物自碳点分离(表4)。
下文表3说明通过药物释放试验所获得的键联基团-药物构筑体半衰期。
表3
a 50%药物自键联基团或粒子中释放的时间。通过HPLC测定。
下文表4说明NDC稳定性数据。
表4
a 25mM乙酸钠缓冲液
b 50mM磷酸盐缓冲液
c 10mM谷胱甘肽(减少的)在50mM磷酸盐缓冲液中
d DEM,无血清,细胞处理后18小时
通过处理H1650细胞,随后在EGFR中进行磷酸-Tyr168的西方墨点检测,且与吉非替尼进行比较来评估碳点-(Cy5)-PEG-Phe-Arg-dPEG2APdMG和碳点-(Cy5)-PEG-Phe-Lys-PABC-APdMG的生物活性。将血清饥饿细胞与每种化合物一起培育达18小时时间段,随后进行EGF刺激。吉非替尼控制显示EGFR的Tyr168磷酸化中的剂量依赖性降低,其在1μM处具有完全剥蚀(图7)。NDCs 6和7同样显示剂量依赖性抑制;用NDC 6处理的细胞在10μM处显示出磷酸-Tyr168的可检测水平。相比之下,NDC 7显示出良好活性,在100nM处磷酸-Tyr168显著降低,且在1μM NDC浓度下完全剥蚀。考虑到可能发生过早药物释放且导致磷酸-Tyr168EGFR中所观测到的降低的可能性,监测用于此等试验中的NDC的稳定性。通过HPLC分析用NDC 6或7(10μM)处理H1650细胞达18小时的媒质等分试样。因为在媒质中未检测到自由药物且NDC无破损,所以证明两种粒子在此等条件下均为稳定的(表3)。除NDC 6和7外,还研究了用于最终活体内研究的二级成像形态的併入。合成具有Phe-Arg-dPEG2-D-Tyr-氨基丙基-dMG的键联基团-药物构筑体,其将D-酪氨酸残基与药物组份结合来连接放射性标记(化合物23和24)。制备NDC碳点-(Cy5)-PEG-Phe-Arg-dPEG2-D-Tyr-APdMG,且在>90%放射化学纯度下用131I成功地进行放射性碘标记(图8)。试剂:
购自商业来源的溶剂和试剂不经进一步纯化即使用。乙腈、二乙醚、二甲基甲酰胺(DMF)、乙酸乙酯、己烷、六氟异丙醇(HFIP)、甲醇、二氯甲烷(DCM)和三氟乙酸(TFA)获自费希尔(Fisher)。二甲亚砜(DMSO)、二异丙基乙胺(DIEA)、三乙胺(TEA)、碳酸钾、N-(叔丁氧基羰基)-氨基丙基溴化物、(3-氨基丙基)三乙氧基硅烷(APTES)、(3-巯基丙基)三甲氧基硅烷(MPTMS)、正硅酸四甲酯(TMOS)、牛胰蛋白酶和购自西格玛-奥德里奇(Sigma-Aldrich)。O-去吗啉基丙基吉非替尼获自多伦多研究化工(Toronto Research Chemicals,TRC)。2-(7-氮杂-1H-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐(HATU)购自金斯瑞(Genescript)。氯三苯甲基-树脂和受保护的氨基酸(Fmoc-Arg-OH、Fmoc-Lys(Mtt)-OH、Fmoc-Phe-OH)获自EMD化工(EMD Chemicals)。Fmoc-N-dPEG2OH、Mmt-S-dPEG8-OH、mal-dPEG12-NHS购自量子生物科学(Quanta Biosciences)。Cy5顺丁烯二酰亚胺和休珀代克斯(Superdex)200(制备级)获自通用生命科学(GE Life Sciences)。DMSO-d和CDCl3购自剑桥同位素(Cambridge Isotopes)。在获自托维克(Torviq)的聚丙烯烧结针筒中进行固相合成。二氧化硅、TLC板、4g、12g、24g和40g RediSep Rf正相滤筒获自特利丹伊斯科(Teledyne ISCO)。
快速色谱:
在特利丹伊斯科CombiFlash Rf上使用4g、12g、24g和40g滤筒进行正相(硅胶)纯化。
分析型HPLC:
在Waters Alliance HPLC系统或Autopure LCMS系统(2767样品管理器、2996光二极管阵列检测器、2420ELS检测器、Micromass ZQ、2525二元梯度模块、管柱流体整理器、515HPLC泵、泵控制模块II)上,在C4或C18 4.6x 50mm反相XBridge分析柱(Waters)上使用5%到95%乙腈于水中(0.5%TFA)的线性梯度以1.2mL/min使样品运行达10分钟。在348nm或650nm处分析样品。
制备型HPLC:
在Waters制备性系统(2996光二极管阵列检测器、2545二元梯度模块)或Autopure LCMS系统上,在C18 19x 150mm反相XBridge制备性管柱(Waters)上使用5%到95%乙腈于水中(0.5%TFA)的线性梯度以20mL/min纯化样品达30分钟。在220或348nm处分析样品。
核磁共振(NMR):
在Bruker Ultrashield 500加上获得1H-NMR和13C-NMR数据。
使用胰蛋白酶的药物释放试验:
用25μM NDC(例如,6或7)或自由键联基团-药物(例如,3、4或5)和200nM胰蛋白酶在37℃下在25mM磷酸盐缓冲液(7.2pH值)中进行试验。为了分析,在指定时间点(例如,5、15、30、60、120分钟或更长)去除70μL份且用淬酸(HCl)灭,随后在HPLC/LCMS上操作。在4℃下将NDC储存在水中。如下制备胰蛋白酶储备液:将1mg胰蛋白酶溶解在1mL水中,等分,随后紧接着进行快速冻结且储存在-80℃下直至四周。在药物释放试验之前,使用Z-Arg-Arg-对硝基-苯胺基质测试酶活性。对于键联基团-药物构筑体或NDC,自由药物%(图3和S3)为自由药物量除以负载药物的初始量。通过对应于348nm处释放药物的HPLC峰的面积确定自由药物量。对于酶处理之前的键联基团-药物构筑体或NDC,负载药物量为348nm处HPLC峰的面积。因为碳点-(Cy5)-PEG-mal在348nm处具有背景吸光度,所以NDC的背景减除为必要的。使用超纯水(18MΩ-cm电阻率)制备所有缓冲剂和溶液。
使用组织蛋白酶B的药物释放试验:
用25μM NDC(例如,6或7)或自由键联基团-药物(例如,3、4或5)和200nM组织蛋白酶B,在37℃下在25mM乙酸钠缓冲液(5.0pH值)中进行试验。此试验不使用DTT。为了分析,在指定时间点(例如,5、15、30、60、120分钟或更长)去除70μL份且用淬酸(HCl)灭,随后在HPLC/LCMS上操作。在4℃下将NDC储存在水中。如下制备组织蛋白酶B储备液:经1mg组织蛋白酶B溶解在1mL 50mM乙酸钠和2.5mM EDTA中,等分,随后紧接着进行快速冻结且储存在-80℃下达数周。在药物释放试验之前,使用Z-Arg-Arg-对硝基-苯胺基质测试酶活性。如前述段落中所述,对于键联基团-药物构筑体或NDC,自由药物%(图4A、4B、6A和6B)为自由药物量除以负载药物的初始量。使用超纯水(18MΩ-cm电阻率)制备所有缓冲剂和溶液。
NDC稳定性试验:
用7.5μM NDC(例如,6或7)在25mM乙酸钠缓冲液(5.0pH值)或50mM磷酸盐缓冲液(7.2pH值)中进行试验,且在37℃下培育直至48小时。同样评估磷酸盐缓冲液(7.2pH值)中的10mM谷胱甘肽(减少的)。随后通过HPLC分析20μL等分试样。对于一个实验,在无血清DEM中用10μM NDC处理H1650细胞(参见下文磷酸EGFR试验)达18小时后,回收且通过HPLC分析媒质。
使用H1650细胞的磷酸EGFR试验:
用2mL10%FBS DEM媒质将H1650细胞(150万个细胞)接种到6孔板中,且使其生长24小时。用1mL无血清DEM媒质洗涤细胞,随后在指定浓度下与吉非替尼或NDC一起培育隔夜(18小时)。随后用50ng/mL EGF处理细胞达5分钟,随后用1mL PBS洗涤。将胰蛋白酶(0.5ml,0.25%)添加到每个孔中,且培育直到细胞分离(约5min)。将1mL10%FBS DEM媒质添加到孔中,且将细胞转移到含有10mL 10%FBS DEM媒质的15mL圆锥形试管中。在4℃下以3000rpm旋转减慢细胞达5分钟。用1mL冷PBS洗涤细胞团,将其转移到1.5mL试管中,且旋转减慢。倾析PBS,且将70μL RIPA(含有蛋白酶和磷酸酶抑制剂)添加到团块中,研磨成粉,且在冰上培育达10分钟。在4℃下以最大速度旋转试管达10分钟。将裂解物转移到新1.5ml试管中且储存在-80℃下。通过布拉德福(Bradford)试验测定蛋白质浓度。在生命技术(Life Technologies)设备上,使用Novex 8%三-甘氨酸凝胶(1.5毫米X 15孔)、三-甘氨酸SDS操作缓冲液、NuPAGE转移缓冲液、0.1%Tween-20于1x TBS洗涤缓冲液和5%牛奶于洗涤缓冲液中作为阻断缓冲液。如下应用初级抗体:抗磷酸化-EGFR(pEGFR,Tyr1068)(1:1000稀释;赛信通(CellSignaling))、抗EGFR(D38B1)(1:5000稀释;赛信通)、单克隆抗β-肌动蛋白克隆株AC-15(1:5000稀释;西格玛-奥德里奇)。应用的二级抗体为山羊抗小鼠IgG-HRP(1:10000稀释;圣克鲁兹生物技术(Santa Cruz Biotechnology))和山羊抗兔IgG-HRP(1:5000稀释;圣克鲁兹生物技术)。
制备O-去吗啉基-吉非替尼,dMG(8,图12(流程3)中):
商业上获得化合物。
1H-NMR(500MHz,DMSO-d6):δ9.69(s,1H),9.47(s,1H),8.48(s,1H),8.22(dd,J=6.9,2.7Hz,1H),7.84(ddd,J=9.1,4.4,2.7Hz,1H),7.78(s,1H),7.41(t,J=9.1Hz,1H),7.22(s,1H),3.98(s,3H)。13C-NMR(500MHz,DMSO-d6):δ155.83,153.91,151.89,146.73,146.20,122.74,121.69,116.50,116.33,109.51,107.18,105.25,55.92。C15H11ClFN3O2的ESI-MS(m/z)(准确质量319.1):[M+H]+计算值320.1,观测值320.2。
合成N-(叔丁氧基羰基)-氨基丙基-dMG(9,图12(流程3)中):
将500mg化合物8溶解在100mL无水DMF中。将K2CO3(mg,mmol)添加到溶液中。在60℃下进行反应达16小时,且通过TLC和/或HPLC检验。在真空中去除溶剂,留下棕色油状物,在24g RediSep Rf正相滤筒上使用20%MeOH于DCM中的DCM线性梯度进行快速纯化。分离最终产物呈白色固体状(312mg,62%产率)。
1H-NMR(500MHz,DMSO-d6):δ9.57(s,1H),8.49(s,1H),8.12(dd,J=6.8,2.7Hz,1H),7.81(m,1H),7.44(t,J=9.1Hz,1H),7.20(s,1H),6.92(t,J=5.7Hz,1H),4.17(t,J=6.0Hz,2H),3.95(s,3H),3.15(q,J=6.5Hz,2H),1.95(p,J=6.5Hz,2H),1.38(s,9H)。13C-NMR(500MHz,DMSO-d6):δ156.01,155.59,154.39,152.61,146.92,123.40,122.29,118.62,116.54,116.37,107.23,102.59,77.51,66.69,55.84,37.22,35.75,28.95,28.22。C23H26ClFN4O4的ESI-MS(m/z)(准确质量476.2):[M+H]+计算值477.2,观测值477.3。
制备氨基丙基-O-去吗啉基-吉非替尼,APdMG(1,图1A和图12(流程3)中)
用1mL TFA:水(9:1)处理化合物9(100mg,0.21mmol)达30min。在真空中去除TFA:水,留下淡黄色油状物。用二乙醚洗涤油状物,随后将其溶解在水:乙腈(1:1)溶液中,冻结且冻干。获得茶色固体(TFA盐,98mg,95%产率)。
1H-NMR(500MHz,DMSO-d6):δ10.73(s,1H),8.80(s,1H),8.03(s,1H),8.05-8.00(m,1H),7.88(s,3H),7.72(ddd,J=9.0,4.3,2.6Hz,1H),7.54(t,J=9.0Hz,1H),7.35(s,1H),4.27(t,J=5.9Hz,2H),4.00(s,3H),3.04(p,J=6.7,6.3Hz,2H),2.13(dt,J=12.2,6.0Hz,2H)。13C-NMR(500MHz,DMSO-d6):δ158.02,148.80,116.96,116.79,107.60,103.66,66.17,56.42,36.39,26.59。C18H18ClFN4O2的ESI-MS(m/z)(准确质量376.1):[M+H]+计算值377.1,观测值377.2。
制备9-芴基甲氧羰基-N-酰胺基-dPEG2-氨基丙基-dMG,Fmoc-dPEG2APdMG(10,图13(流程4)):
制备含有dPEG2APdMG 2(25mg,0.05mmol,TFA盐)和Fmoc-N-酰胺基-dPEG2-COOH(20mg,0.05mmol)于DMF(500μL)中的溶液。向DMF(100μL)中添加DIEA(19mg,0.15mmol,26μL),随后添加HATU溶液(19mg,0.05mmol)。在室温下进行反应达30min,通过LCMS完全测定。在真空中减小体积,且通过硅胶色谱法,使用乙酸乙酯梯度和10%甲醇于乙酸乙酯中来进行纯化。收集部分,合并且在真空中去除溶剂。分离产物为白色固体(84%产率)。
1H-NMR(500MHz,DMSO-d6):δ9.55(d,J=10.7Hz,1H),8.51(d,J=2.4Hz,1H),8.18-8.07(m,1H),7.96(t,J=5.5Hz,1H),7.87(d,J=7.6Hz,2H),7.83-7.76(m,2H),7.67(d,J=7.5Hz,2H),7.49-7.36(m,3H),7.31(td,J=7.5,1.2Hz,3H),7.21(s,1H),4.28(d,J=6.9Hz,2H),4.24-4.10(m,3H),3.94(s,3H),3.60(t,J=6.4Hz,2H),3.46(s,4H),3.36(t,J=6.0Hz,2H),3.27(q,J=6.6Hz,2H),3.10(q,J=5.9Hz,2H),2.32(t,J=6.5Hz,2H),1.97(p,J=6.5Hz,2H)。13C-NMR(500MHz,CDCl3):δ170.08,143.85,127.54,126.98,125.10,120.05,102.60,69.42,69.04,66.78,66.58,55.87,46.69,40.01,39.94,39.85,39.77,39.68,39.60,39.51,39.43,39.35,39.25,39.18,39.07,39.01,36.16,35.69,28.67,0.08。C40H41ClFN5O7的ESI-MS(m/z)(准确质量757.27):[M+H]+计算值758.3,观测值758.4。
制备氨基-dPEG2-氨基丙基-dMG,dPEG2APdMG(2,图1A和图13(流程4)中):
将化合物10(10mg,0.013mmol)溶解在含30%哌啶的DMF(1mL)中,且使其在室温下反应达15min。在真空中去除溶剂,将其溶解在水/乙腈中,且通过反相(C18)HPLC进行纯化。回收产物呈白色粉末状(7mg,80%产率)。
1H-NMR(500MHz,DMSO-d6):δ10.66(s,1H),8.78(s,1H),8.05-7.97(m,3H),7.80(s,3H),7.72(ddd,J=9.0,4.3,2.6Hz,1H),7.54(t,J=9.1Hz,1H),7.32(s,1H),4.20(t,J=6.1Hz,2H),4.00(s,3H),3.65-3.48(m,7H),3.27(q,J=6.6Hz,2H),2.96(q,J=5.5Hz,2H),2.34(t,J=6.5Hz,2H),1.98(t,J=6.5Hz,2H)。13C-NMR(500MHz,DMSO-d6):δ116.96,69.55,69.29,66.73,66.61,56.38,38.57,36.03,35.63,28.58。C25H31ClFN5O5的ESI-MS(m/z)(准确质量535.20):[M+H]+计算值536.2,观测值536.3。
制备Boc-N-氨基-(dPEG2)3-Phe-Arg(Pbf)-OH(11,图14(流程5)中):
将氯三苯甲基树脂(100mg,0.1mmol,1mmol/g)转移到烧结注射器反应器中,且使其悬浮于2mL无水DCM中达10min。施配溶剂,且将DIEA于无水DCM中的溶液、随后Fmoc-Arg(Pbf)-OH(97.5mg,1.5当量)于无水DCM中的溶液抽取到注射器中;且搅拌40min。施配溶液,且用DCM 2次洗涤树脂2min,随后用DMF洗涤2次。进行标准固相肽合成程序以获得最终肽。简单地说,通过使用30%哌啶/DMF(1mL)2次洗涤树脂10min来实现Fmoc去除保护基。随后用DMF(1mL)洗涤4次各2min。使用3当量过量的受保护氨基酸(于2mL DMF中)、9当量过量DIEA(120mg,0.9mmol,160μL,于1mL DMF中)、3当量过量HATU(mmol,mg,μL,于2mL DMF中),以所述顺序将其添加到注射器中,且摇晃1小时,以在室温下进行偶合反应。随后用DMF(1mL)洗涤4次各2min。添加Fmoc-Phe(116mg),随后添加Fmoc-N-dPEG2-OH的三种残基(120mg)。序列完整后,进行最终Fmoc去除保护基且洗涤,使用BOC酐(mmol,mg)和DIEA(mmol,mg,μL)于2mL DMF中来封盖N端胺。用DMF(1mL,2min,2次)洗涤肽-树脂,随后用DCM(1mL,2min,4次)洗涤。随后通过将50%HFIP于DCM中(2mL)添加到注射器中且在室温下摇晃1小时,使受保护肽产物裂解脱离树脂。随后通过反相HPLC纯化粗产物肽。C54H86N8O17S的ESI-MS(m/z)(准确质量1150.56):[M+H]+计算值1151.6,观测值1151.7。
制备Boc-N-酰胺基-(dPEG2)3-Phe-Arg(Pbf)-APdMG(12,图14(流程5)中):
制备含有化合物11(23mg,0.02mmol,1当量)和APdMG 1(9mg,0.024mmol,1.2当量)于DMF(1mL)中的溶液。向其中添加DIEA(10mg,0.08mmol,14μL,4当量),随后添加HATU(9mg,0.024mmol,1.2当量)。通过HPLC监测反应,且反应在30分钟内完成。在真空中去除溶剂,随后在DCM中进行再悬浮。用水洗涤DCM溶液4次,随后蒸发留下茶色油状物。C72H102ClFN12O18S的ESI-MS(m/z)(准确质量1508.68):[M+H]+计算值1509.7,观测值1509.7;[M+2H]2+计算值755.4,观测值755.0。
制备Boc-N-酰胺基-(dPEG2)3-Phe-Arg(Pbf)-dPEG2APdMG(13,图14(流程5)中):
制备含有化合物11(30mg,0.026mmol,1当量)和dPEG2APdMG 2(18mg,0.034mmol,1.3当量)于DMF(1mL)中的溶液。向其中添加DIEA(14mg,0.1mmol,18μL,4当量),随后添加HATU(13mg,0.034mmol,1.3当量)。通过HPLC监测反应,且反应在30分钟内完成。在真空中去除溶剂,随后在DCM中进行再悬浮。用水洗涤DCM溶液4次,随后蒸发留下茶色油状物。C79H115ClFN13O21S的ESI-MS(m/z)(准确质量1667.77):[M+2H]2+计算值834.9,观测值834.7。
制备H2N-(dPEG2)3-Phe-Arg-APdMG(14,流程6中):
将化合物12(自前述步骤为约0.02mmol)添加到TFA/水(9:1,1mL)中,且在室温下静置1小时。蒸发反应物,随后将其溶解在ACN/水中,冻结且冻干,留下茶色固体。通过反相HPLC纯化粗物质。留下呈白色固体状的最终产物(14mg)。C54H78ClFN12O13的ESI-MS(m/z)(准确质量1156.55):[M+H]+计算值1157.6,观测值1157.8;[M+2H]2+计算值579.3,观测值579.1。
制备H2N-(dPEG2)3-Phe-Arg-(dPEG2)-APdMG(15,图14(流程5)中):
将化合物13(自前述步骤为约0.026mmol)添加到TFA/水(9:1,1mL)中,且在室温下静置1小时。蒸发反应物,随后将其溶解在ACN/水中,冻结且冻干,留下茶色固体。通过反相HPLC纯化粗物质。留下呈白色固体状的最终产物(24mg)。C61H91ClFN13O16的ESI-MS(m/z)(准确质量1315.64):[M+H]+计算值1316.7,观测值1316.5;[M+2H]2+计算值658.6,观测值658.5。
制备S-乙酰基-巯基乙酰胺基-(dPEG2)3-Phe-Arg-APdMG(16,图14(流程5)中):
制备含有化合物14(5mg,0.004mmol,1当量)和DIEA(1.5mg,0.012mmol,2μL,3当量)于DMF(200μL)中的溶液。随后将SAMA-OPfp(2mg,0.006mmol,1.5当量)于DMF(100μL)中添加到溶液中,且使其反应达1小时。在真空中去除溶剂,随后通过反相HPLC纯化。回收3mg白色固体。C61H91ClFN13O16的ESI-MS(m/z)(准确质量1315.64):[M+H]+计算值1316.7,观测值1316.5;[M+2H]2+计算值658.6,观测值658.5。
制备S-乙酰基-巯基乙酰胺基-(dPEG2)3-Phe-Arg-dPEG2APdMG(17,图14(流程5)中):
制备含有化合物15(5mg,0.004mmol,1当量)和DIEA(1.5mg,0.012mmol,2μL,3当量)于DMF(200μL)中的溶液。随后将SAMA-OPfp(2mg,0.006mmol,1.5当量)于DMF(100μL)中添加到溶液中,且使其反应达1小时。在真空中去除溶剂,随后通过反相HPLC纯化。回收3mg白色固体。C65H95ClFN13O18S的ESI-MS(m/z)(准确质量1431.63):[M+2H]2+计算值716.8,观测值716.7。
制备巯基乙酰胺基-(dPEG2)3-Phe-Arg-APdMG,Phe-Arg-APdMG(3,图1B和图14(流程5)中):
紧接在使用之前进行此步骤。将1mg化合物16溶解在100μL水/MeOH(1:1)中,向其中添加2μL 1N NaOH。15分钟后,添加2μL 1M HCl来中和。直接使用溶液。C56H80ClFN12O14S的ESI-MS(m/z)(准确质量1230.53):[M+H]+计算值1231.5,观测值1231.4;[M+2H]2+计算值616.3,观测值616.3。
制备巯基乙酰胺基-(dPEG2)3-Phe-Arg-dPEG2APdMG,Phe-Arg-dPEG2APdMG(4,图1C和图14(流程5)中):
紧接在使用之前进行此步骤。将1mg化合物17溶解在100μL水/MeOH(1:1)中,向其中添加2μL 1N NaOH。15分钟后,添加2μL 1M HCl来中和。直接使用溶液。C63H93ClFN13O17S的ESI-MS(m/z)(准确质量1389.62):[M+H]+计算值1390.6,观测值1390.5;[M+2H]2+计算值695.8,观测值695.7。
制备Fmoc-Lys(Mtt)-PABOH(18,图15(流程6)中):
制备Fmoc-Lys(Mtt)-OH(748mg,1.2mmol,1当量)和对氨基苄醇(300mg,2.4mmol,2当量)于DMF(5mL)中的溶液。添加DIEA(465mg,3.6mmol,630μL,3当量),随后添加HATU(502mg,1.3mmol,1.1当量)于DMF(2mL)中的溶液。反应在30分钟内完成,且通过HPLC/LCMS测定。在真空中部分去除溶剂,且用乙酸乙酯/水进行萃取。用水洗涤乙酸乙酯层4次,随后蒸发产生橙色固体。在40g RediSep Rf正相滤筒上,使用己烷和乙酸乙酯的线性梯度来纯化粗物质。分离最终产物呈白色固体状(850mg,97%产率)。
1H-NMR(500MHz,CDCl3):δ7.96(s,1H),7.73(d,J=7.9Hz,2H),7.54(d,J=7.5Hz,2H),7.47-7.41(m,6H),7.39-7.20(m,12H),7.18-7.11(m,2H),7.05(d,J=7.9Hz,2H),5.29(s,1H),4.63(s,1H),4.44(d,J=6.4Hz,2H),2.28(s,3H),2.11(t,J=6.9Hz,2H),1.88(s,1H),1.58(s,5H),1.51(s,1H),1.38(s,2H)。13C-NMR(500MHz,CDCl3):δ146.36,143.22,141.32,135.70,128.57,128.52,128.49,127.79,127.74,127.12,126.14,124.92,120.16,120.02,77.27,77.02,76.76,70.62,64.92,60.41,47.16,43.30,30.57,23.43,21.07,20.93,14.21,0.01。C48H47N3O4的ESI-MS(m/z)(准确质量729.36):[M+H]+计算值730.4,观测值730.2。
制备Fmoc-Phe-Lys(Mtt)-PABOH(19,图15(流程6)中):
使用9mL含30%哌啶的DMF,自Fmoc-Lys(Mtt)-PABOH 18(425mg,0.6mmol,1当量)中去除Fmoc基团达10分钟。在真空中去除溶剂,且将所得油状物再悬浮于10mLDMF中。制备Fmoc-Lys(Mtt)-OH(748mg,1.2mmol,1当量)和对氨基苄醇(300mg,2.4mmol,2当量)于DMF(5mL)中的溶液。添加DIEA(465mg,3.6mmol,630μL,3当量),随后添加HATU(502mg,1.3mmol,1.1当量)于DMF(2mL)中的溶液。反应在30分钟内完成,且通过HPLC/LCMS测定。在真空中部分去除溶剂,且用乙酸乙酯/水进行萃取。用水洗涤乙酸乙酯层4次,随后蒸发,产生橙色固体。在40g RediSep Rf正相滤筒上,使用己烷和乙酸乙酯的线性梯度来纯化粗物质。分离最终产物呈白色固体状(850mg,97%产率)。
1H-NMR(500MHz,CDCl3)δ8.20(s,1H),7.73(d,J=7.6Hz,2H),7.50(s,2H),7.48-7.34(m,8H),7.34-7.20(m,11H),7.20-7.07(m,7H),7.04(d,J=8.1Hz,2H),6.28(s,1H),5.22(s,1H),4.62(s,2H),4.43(dd,J=10.7,6.7Hz,1H),4.32(d,J=11.5Hz,1H),3.05(s,2H),2.27(s,3H),2.11-2.02(m,2H),1.89(s,1H),1.46(d,J=6.3Hz,1H),1.26(s,2H)。13C-NMR(500MHz,CDCl3)δ146.35,141.31,136.97,135.70,129.10,128.93,128.57,128.50,127.82,127.74,127.71,127.39,127.12,126.14,124.91,124.84,120.10,120.04,77.28,77.23,77.02,76.77,70.60,67.15,64.96,54.06,47.08,43.35,31.30,30.60,23.52,20.93。C57H56N4O5的ESI-MS(m/z)(准确质量876.43):[M+H]+计算值877.4,观测值877.3。
制备Fmoc-Phe-Lys(Mtt)-PABC-APdMG(21,图15(流程6)中):
将化合物19(420mg,0.5mmol,1当量)溶解在无水DCM(20mL)中。添加吡啶(216mg,2.7mmol,5.4当量),随后添加氯甲酸4-硝基苯酯(180mg,0.9mmol,1.8当量)于无水DCM中的溶液。在室温下进行反应达2小时,随后通过HPLC和TLC检验。在真空中去除溶剂,随后通过在24g RediSep Rf正相滤筒上使用己烷和乙酸乙酯的线性梯度来进行纯化。分离产物Fmoc-Phe-Lys(Mtt)-PABC-pNP(20)呈黄色固体状(360mg,70%产率)。将Fmoc-Phe-Lys(Mtt)-PABC-pNP(50mg,0.05mmol,1当量)溶解在无水DCM(3mL)中。随后将APdMG 1(25mg,0.05mmol,TFA盐,1当量)与DIEA(65mg,0.5mmol,90μL,10当量)于无水DCM中的溶液添加到Fmoc-Phe-Lys(Mtt)-PABC-pNP中。在室温下进行反应达4小时,随后通过HPLC和TLC检验。在真空中去除溶剂,且在4g RediSepRf正相滤筒上用己烷和乙酸乙酯的线性梯度来纯化粗物质。分离最终产物呈黄色固体状(38mg,60%产率)。C76H72ClFN8O8的ESI-MS(m/z)(准确质量1278.51):[M+H]+计算值1279.5,观测值1279.4;[M+2H]2+计算值640.3,观测值640.3。
制备Mmt-S-dPEG8-Phe-Lys(Mtt)-pABC-dMG(22,图15(流程6)中):
用2mL含30%哌啶的DMF对18mg化合物21(0.014mmol,1当量)进行去除保护基。5分钟后,通过HPLC/LCMS证实反应完全,且在真空中去除溶剂。将所得油状物溶解在DMF(0.5mL)中,向其中添加Mmt-S-dPEG8-COOH(13mg,0.017mmol,1.2当量)和DIEA(9mg,0.070mmol,13μL,5当量)。制备HATU(6mg,0.014mmol,1.2当量)于DMF(200μL)中的溶液,且将其添加到反应物中。1小时后,通过HPLC/LCMS认为反应完全,且在真空中去除溶剂。在4g RediSep Rf正相滤筒上使用10%MeOH于DCM中的DCM线性梯度来快速纯化剩余油状物。分离最终产物呈白色固体状(23mg,92%产率)。C100H114ClFN8O16S的ESI-MS(m/z)(准确质量1768.77):[M+2H]2+计算值885.9,观测值886.0。
制备HS-dPEG8-Phe-Lys-PABC-氨基丙基-dMG(5,图15(流程6)中):
用2mL 0.5%TFA/5%TIS于DCM中处理10mg化合物22(5.6μmol)达2小时,随后通过HPLC/LCMS检验以确认完全去除保护基。在真空中去除溶液,且随后用冷醚洗涤3次。将白色固体溶解在水/乙腈(1:1)中,冻结且冻干。不经进一步纯化即使用所得白色固体(6mg,86%产率)。C60H82ClFN8O15S的ESI-MS(m/z)(准确质量1240.53):[M+H]+计算值1241.5,观测值1241.6;[M+2H]2+计算值621.3,观测值621.3。
制备碳点-(Cy5)-PEG-顺丁烯二酰亚胺:
用氨基硅烷(APTES)于DMSO中使顺丁烯二酰亚胺和NHS酯官能化聚乙二醇(mal-dPEG12-NHS)结合(摩尔比:PEG-NHS:APTES:DMSO为1:0.9:60)。在室温下将反应混合物静置在氮气下达48小时以产生硅烷官能化mal-dPEG(mal-dPEG-APTES)。在DMSO中使顺丁烯二酰亚胺官能化Cy5(mal-Cy5)与硫醇-硅烷(MPTMS)反应(摩尔比:Cy5:MPTMS:DMOS为1:25:1150)。在室温下将反应物静置在氮气下达24小时以产生硅烷官能化Cy5(Cy5-MPTMS)。随后将TMOS和Cy5-MPTMS滴定到氢氧化氨溶液(pH值约为8)中(摩尔比:TMOS:Cy5:NH3:H2O为1:0.001:0.44:1215)。在室温下以600rpm搅拌溶液达24小时,以形成均相Cy5囊封二氧化硅纳米粒子。随后将mal-dPEG-APTES和硅烷官能化聚乙二醇(PEG-硅烷,约500MW,盖勒斯特(Gelest))添加到合成溶液中,以使粒子聚乙二醇化和表面官能化(PEG-硅烷:TMOS:mal-PEG-APTES为1:2.3:0.006)。在室温下以600rpm搅拌溶液达24小时,随后在不搅拌的情况下在80℃下再培育24小时。用去离子水以2000mL透析溶液达两天(10k MWCO),用200nm注射器过滤器过滤,且最后进行色谱纯化(休珀代克斯200),产生所要mal-碳点。
制备碳点-(Cy5)-PEG-Phe-Arg-dPEG2-Gly-D-Tyr-APdMG:
使用用于获得化合物6的相同整体合成策略。合成键联基团-药物构筑体Phe-Arg-dPEG2-Gly-D-Tyr-APdMG(23)。C74H105ClFN15O20S的(ESI-MS(m/z)(准确质量1609.71):[M+2H]2+计算值805.9,观测值805.6)。如针对NDC 6和7所描述,此构筑体连接到碳点-(Cy5)-PEG-mal上。
制备碳点-(Cy5)-PEG-Phe-Lys-PABC-Gly-D-Tyr-APdMG:
使用用于获得化合物7的相同整体合成策略。合成键联基团-药物构筑体Phe-Lys-PABC-Gly-D-Tyr-APdMG(24)。C71H94ClFN10O18S的(ESI-MS(m/z)(准确质量1460.61):[M+H]+计算值1461.6,观测值1461.3;[M+2H]2+计算值731.3,观测值731.如针对NDC 6和7所描述,此构筑体连接到碳点-(Cy5)-PEG-mal上。
制备放射性碘化碳点-(Cy5)-PEG-Phe-Arg-dPEG2-Gly-D-Tyr-氨基丙基-APdMG:
使用Iodogen协定对碳点-(Cy5)-PEG-Phe-Arg-dPEG2-Gly-D-Tyr-APdMG进行放射性碘化。在PD10管柱上纯化碘化反应物,随后通过GPC(Superdex)分析。
- 还没有人留言评论。精彩留言会获得点赞!