基于动态加权相互作用网络中关键蛋白质识别方法与流程
本发明属于生物信息技术领域,主要是在蛋白质相互作用网络中通过动态加权相互作用网络识别关键蛋白质的技术,特别涉及在动态加权ppi网络中通过网络拓扑特性和蛋白质生物属性识别关键蛋白质的方法。
背景技术:
在生物细胞中,关键蛋白质是实现细胞功能不可缺少的,关键蛋白质的探测有助于理解细胞代谢,生长和发育的规律。因此,关于关键蛋白质的识别成为蛋白质组学领域的一项重要研究工作之一。虽然目前对于蛋白质相互作用网络中的关键蛋白质的识别取得了一些成果,但由于生命系统的高度复杂性和随机性,其他领域中卓有成效的方法往往在ppi网络分析中不一定取得理想的效果,忽略了蛋白质相互作用网络的动态性,导致识别出来的蛋白质准确率较低。
在本发明作出之前,在已有的方法中,关键蛋白质识别方法都是根据ppi中的某一顶点的某种重要性指标,如顶点的各种中心度等来排序,取其中较大的k个作为关键蛋白质。然而,这类重要性指标往往很多,如度的中心度、介度中心度、聚类中心度等,这样识别关键蛋白质的缺点是:(1)对于某一蛋白质而言,其某种中心度较高,而其他中心度未必也很高,这将导致识别的关键蛋白质精确度低;(2)基于蛋白质拓扑特性的关键蛋白质预测方法,不但依赖于蛋白质相互作用网络的准确性,同时缺乏考虑关键蛋白质的生物特性。(3)仅仅考虑了静态ppi网络下蛋白质的一些特性,忽略了蛋白质相互作用网络随着时间不断发生变化的。
技术实现要素:
本发明的目的就在于克服上述缺陷,研制基于动态加权相互作用网络中关键蛋白质识别方法。
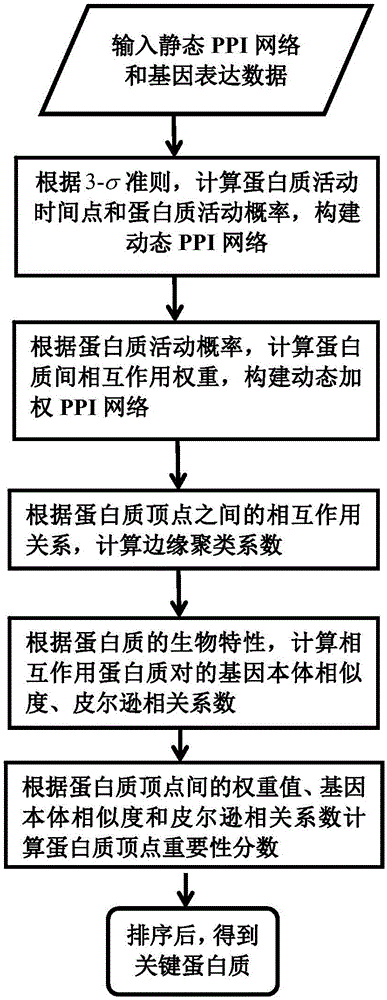
基于动态加权相互作用网络中关键蛋白质识别方法使用3-σ准则,计算蛋白质活动时间点和蛋白质活动概率,构建动态ppi网络,然后,根据蛋白质活动概率,计算蛋白质间相互作用权重,构建动态加权ppi网络。在建立好的动态加权ppi网络上,根据蛋白质网络的拓扑特性和生物属性,计算相互作用蛋白质对间的边缘聚类系数、基因本体相似度和皮尔逊相关系数。之后,得到一个重要性分数,最终按分值由大到小排列,输出分值对应的k个蛋白质即为最后结果。
基于动态加权相互作用网络中关键蛋白质识别方法,其主要技术特征在于如下步骤:
(1)输入ppi网络和基因表达数据;
(2)根据3-σ准则,计算蛋白质活动时间点和蛋白质活动概率,构建动态ppi网络;
(3)根据蛋白质活动概率,计算蛋白质间相互作用权重,构建动态加权ppi网络;
(4)根据蛋白质顶点之间的相互作用关系,计算边缘聚类系数;
(5)根据蛋白质的生物特性,计算相互作用蛋白质对的基因本体(geneontology)相似度、皮尔逊相关系数(personcorrelationcoefficient);
(6)根据蛋白质顶点间的权重值、基因本体相似度和皮尔逊相关系数计算蛋白质顶点重要性分数;
(7)获得每个顶点的重要性分数并由大到小排序,排序后最大的k个值为关键蛋白质。
进一步地,所述步骤(2)根据3-σ准则,计算蛋白质活动时间点和蛋白质活动概率,构建动态ppi网络:通过步骤(1)输入ppi网络和基因表达数据,每个蛋白质具有m个时间点,每个时间点对应了一个基因表达值,根据基因表达值的平均值和标准差,计算活动阈值。将活动阈值与每个蛋白质的基因表达值进行比较,构建动态ppi网络。
进一步地,所述步骤(3)根据蛋白质活动概率,计算蛋白质间相互作用权重,构建加权动态ppi网络:通过步骤(2)计算蛋白质活动概率,为每对活动的相互作用蛋白质赋予一个权重,结合动态ppi网络,构建动态加权ppi网络。
进一步地,所述步骤(6)根据蛋白质顶点间的权重值、基因本体相似度和皮尔逊相关系数计算蛋白质顶点重要性分数:整合相互作用蛋白质网络的拓扑特性以及蛋白质的生物属性,建立一个蛋白质重要性计算公式,得出每个蛋白质的最终得分。
本发明的优点和效果在于该方法考虑了蛋白质网络的动态性,整合了蛋白质相互作用网络的拓扑特性以及蛋白质的生物属性信息,进而克服数据的不完全性、动态性以及数据的噪声性所带来的负面影响。在动态加权ppi网络上,将蛋白质相互作用网络的拓扑特性与蛋白质生物属性相结合有助于提高识别关键蛋白质的准确性,提高了关键蛋白质识别的效率,扩展了该技术在生物信息领域的应用范围和实用性。
附图说明
图1——本发明在动态加权相互作用网络中关键蛋白质识别方法流程示意图;
图2——本方法(iep-dppi)和其他方法在静态ppi网络和动态ppi网络中识别的关键蛋白质数量比较图。
具体实施方式
本发明的技术思路是:
根据静态ppi网络和基因表达数据,通过3-σ准则,计算蛋白质活动时间点和蛋白质活动概率,构建动态ppi网络,然后,根据蛋白质活动概率,计算蛋白质间相互作用权重,构建动态加权ppi网络。基于动态加权ppi网络,根据蛋白质顶点之间的相互作用关系,计算边缘聚类系数;根据蛋白质的生物特性,计算相互作用蛋白质对的基因本体(geneontology)相似度、皮尔逊相关系数(personcorrelationcoefficient)。最后,根据蛋白质顶点间的权重值、基因本体相似度和皮尔逊相关系数计算蛋白质顶点重要性分数,最终获得每个顶点的重要性分数并由大到小排序,排序后最大的k个值为关键蛋白质。在动态加权ppi网络中,整合生物属性和拓扑特性有助于理解未知蛋白质的功能,对于解释特定功能的分子机制有着重要意义,同时能够对药物靶细胞设计等提供重要的理论依据。所以基于动态加权相互作用网络中关键蛋白质识别方法很自然地适用于关键蛋白质的探测。
下面结合附图和具体实施方式对本发明进行详细说明。
步骤1:输入静态ppi网络和基因表达数据
步骤2:根据3-σ准则,计算蛋白质活动时间点和蛋白质活动概率,构建动态ppi网络
基因表达数据通常由m个时间点表达谱组成。设gei(p)表示为在时间点i基因p的基因表达值。设u(p)和σ(p)分别表示基因表达数据的算术平均值和标准差。设x是正态分布的实数随机变量n(u,σ2)。对于任何k>0,p{|x-u|<kσ}=2φ(k)-1其中,φ(·)是标准正规定律的分布函数。特别是,当k=1,2,3时,p{|x-u|<σ}=p{u-σ<x<u+σ}≈0.6827,p{|x-u|<2σ}≈0.9545和p{|x-u|<3σ}≈0.9973。并给出如下公式:
使用3-σ准则计算不同时间点的k-σ阈值,公式如下:
使用pri来表示蛋白质在i时间点的活动概率。因此,使用四个水平来表示基于3-σ准则的时间点i蛋白质的活动概率,其通过公式(4)计算。
首先基于每种蛋白质的活动概率构建动态蛋白质相互作用网络。对于蛋白质p,当基因表达值大于active_threshk(p)时,认为它在时间点i是活动的。蛋白质活动的时间点的集合可以表示为tp={t1,t2,...,tm},1≤m≤36。如果蛋白质p和蛋白质q在静态网络上相互作用并且具有共同的活动时间点,则认为蛋白质p和q在动态ppi网络上彼此相互作用。cot(p,q)=1表明蛋白质p和q在动态ppi网络上具有相互作用。
步骤3:根据蛋白质活动概率,计算蛋白质间相互作用权重,构建动态加权ppi网络,通过公式(6)可以有效地给出时间点i的边(p,q)的加权值:
weight(ei(p,q))=pri(p)·pri(q)(6)
其中ei(p,q)表示p和q之间的边在时间点i有相互作用。
动态ppi网络的活动水平由每条边的权重之和计算得出,其给出如下:
基于公式(7),每条边的加权值平均值计算如下:
其中|t|表示蛋白质p和q的共同活动时间点的数量。
步骤4:根据蛋白质顶点之间的相互作用关系,计算边缘聚类系数
边(u,v)的边聚类系数可以通过以下公式计算得出:
其中,nu和nv分别表示顶点u和v邻居节点的集合。du和dv分别代表顶点u和v的度。
步骤5:根据蛋白质的生物特性,计算相互作用蛋白质对的基因本体(geneontology)相似度、皮尔逊相关系数(personcorrelationcoefficient)
(1)基因本体(geneontology)相似度
geneontology包含生物过程、细胞组成和分子功能,广泛用于检测基因功能间的关联。geneontology相似度定义如下:
其中,term(g1)和term(g2)分别表示g1和g2的geneontology术语数。
(2)皮尔逊相关系数(personcorrelationcoefficient)
皮尔逊相关系数(personcorrelationcoefficient)用于测量共表达的蛋白质对,这是两个线性依赖变量之间相关强度广泛使用的度量。基因x和y的皮尔逊相关系数定义为:
其中,m是基因表达数据的样品数,g(x,i)和g(y,i)分别代表样品i中基因x和y的表达水平。
步骤6:根据蛋白质顶点间的权重值、基因本体相似度和皮尔逊相关系数计算蛋白质顶点重要性分数
定义每个相互作用边(u,v)的重要性性分数。蛋白质对u和v的必要性评分如下:
esps(u,v)=αecc+βsum_ave+ηpcc+γsimmatch(12)
其中,α,β,γ,η∈(0,1)
对于蛋白质v,espsi(v)定义为重要性得分的总和。
espsi(u)=∑v∈n(u)esps(u,v)(13)
其中,n(u)是节点u的邻居节点的集合。
步骤7:获得每个顶点的重要性分数并由大到小排序,排序后最大的k个值为关键蛋白质。
实施例:
在dip数据集中将本发明提出的方法(iep-dppi)与dc,lac,sc,bc,nc现有的方法在静态ppi网络和动态ppi网络中进行比较。对于每种方法,本发明选择前100到前600个蛋白质结果作为候选集。
dip数据集的预测结果如图2所示。本发明提出的方法iep-dppi可以比其他方法在识别关键蛋白质方面获得更好的结果。同时,基于动态ppi网络预测关键蛋白质的每个典型中心度量的表现优于原始静态ppi网络。当检测到前600种蛋白质时,很明显iep-dppi方法在静态ppi网络上识别出的关键蛋白质的数量比bc方法高55%。
- 还没有人留言评论。精彩留言会获得点赞!